A critical part of the MNK and WND physiological response are their ability to change intracellular localisation in response to copper levels, relocating to sites where copper transport is required.
Under basal physiological copper levels, MNK and WND concentrate within the trans Golgi network (TGN) region, where they are postulated to pump copper into the TGN lumen for incorporatio into proteins on the secretory pathway.
When copper levels are raised, MNK has been reported to traffic to the plasma membrane and to the basolateral membrane in some polarized cell.
In response to elevated copper levels WND traffics to sub apical vesicles in some polarized cell lines, and has also been deonstrated partially at the apical membrane.
When intracellular copper levels are reduced, both transporters return via an endocytic route to the TGN.
Tuesday, 20 November 2007
My project (12)
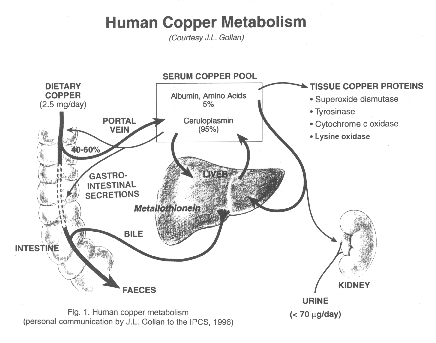
Human copper metabolism
Ingested copper is absorp from the gut 40-50%.
Copper bind to protein (mostly ceruloplasmin 95%).
Copper is transported to liver. Copper is secreted to bile and excreted to the gut again in feces
My project (11)
There are 13 types of ATP-ase
Na+/K+ transporting: ATP1A1, ATP1A2, ATP1A3, ATP1A4, ATP1B1, ATP1B2, ATP1B3, ATP1B4
Ca++ transporting: ATP2A1, ATP2A2, ATP2A3, ATP2B1, ATP2B2, ATP2B3, ATP2B4, ATP2C1
Mg++ transporting: ATP3
H+/K+ exchanging: ATP4A, ATP4B
H+ transporting, mitochondrial: ATP5A1, ATP5B, ATP5C1, ATP5C2, ATP5D, ATP5E, ATP5F1, ATP5G1, ATP5G2, ATP5G3, ATP5H, ATP5I, ATP5J, ATP5J2, ATP5L, ATP5L2, ATP5O, ATP5S
H+ transporting, lysosomal: ATP6AP1, ATP6AP2, ATP6V1A, ATP6V1B1, ATP6V1B2, ATP6V1C1, ATP6V1C2, ATP6V1D, ATP6V1E1, ATP6V1E2, ATP6V1F, ATP6V1G1, ATP6V1G2, ATP6V1G3, ATP6V1H, ATP6V0A1, ATP6V0A2, ATP6V0A4, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V0D2, ATP6V0E
Cu++ transporting: ATP7A (see also ATP7A), ATP7B (see also ATP7B)
Class I, type 8: ATP8A1, ATP8B1, ATP8B2, ATP8B3, ATP8B4
Class II, type 9: ATP9A, ATP9B
Class V, type 10: ATP10A, ATP10B, ATP10D
Class VI, type 11: ATP11A, ATP11B, ATP11C
H+/K+ transporting, nongastric: ATP12A
type 13: ATP13A1, ATP13A2, ATP13A3, ATP13A4, ATP13A5
Na+/K+ transporting: ATP1A1, ATP1A2, ATP1A3, ATP1A4, ATP1B1, ATP1B2, ATP1B3, ATP1B4
Ca++ transporting: ATP2A1, ATP2A2, ATP2A3, ATP2B1, ATP2B2, ATP2B3, ATP2B4, ATP2C1
Mg++ transporting: ATP3
H+/K+ exchanging: ATP4A, ATP4B
H+ transporting, mitochondrial: ATP5A1, ATP5B, ATP5C1, ATP5C2, ATP5D, ATP5E, ATP5F1, ATP5G1, ATP5G2, ATP5G3, ATP5H, ATP5I, ATP5J, ATP5J2, ATP5L, ATP5L2, ATP5O, ATP5S
H+ transporting, lysosomal: ATP6AP1, ATP6AP2, ATP6V1A, ATP6V1B1, ATP6V1B2, ATP6V1C1, ATP6V1C2, ATP6V1D, ATP6V1E1, ATP6V1E2, ATP6V1F, ATP6V1G1, ATP6V1G2, ATP6V1G3, ATP6V1H, ATP6V0A1, ATP6V0A2, ATP6V0A4, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V0D2, ATP6V0E
Cu++ transporting: ATP7A (see also ATP7A), ATP7B (see also ATP7B)
Class I, type 8: ATP8A1, ATP8B1, ATP8B2, ATP8B3, ATP8B4
Class II, type 9: ATP9A, ATP9B
Class V, type 10: ATP10A, ATP10B, ATP10D
Class VI, type 11: ATP11A, ATP11B, ATP11C
H+/K+ transporting, nongastric: ATP12A
type 13: ATP13A1, ATP13A2, ATP13A3, ATP13A4, ATP13A5


My project (8)
Intracellular levels of copper are in part controlled by two copper-transporting P-type ATPases, ATP7A(Menkes disease protein, MNK) and ATP7B(Wilson disease protein, WND). Both transporters are made up of 8 transmembrane domains and hydrolase ATP to translocate ions across cell membranes. MNK is involved in copper absorption, predominantly in the intestine and the kidney, whilst WND localizes predominantly to the liver and regulates copper clearanceby excreting it into bile.
MENKES DISEASE(#309400)
Clinical Synopsis
INHERITANCE :
X-linked recessive
GROWTH :
Height
Short stature
Other
Intrauterine growth retardation
HEAD AND NECK :
Head
Microcephaly
Brachycephaly
Wormian bones
Face
Pudgy cheeks
CARDIOVASCULAR :
Vascular
Intracranial hemorrhage
SKELETAL :
Osteoporosis
Skull
Wormian bones
Limbs
Metaphyseal widening with spurs
SKIN, NAILS, HAIR :
Skin
Hypopigmentation
Hair
Steely, kinky, sparse hair
Twisted and partial breaks on magnification
NEUROLOGIC :
Central nervous system
Degenerative neurologic disorder with onset age 1 month
Hypertonia
Seizures
Intracranial hemorrhage
Hypothermia
LABORATORY ABNORMALITIES :
Low copper and ceruloplasmin
WILSON DISEASE (#277900)
Clinical Synopsis
INHERITANCE :
Autosomal recessive
HEAD AND NECK :
Eyes
Kayser-Fleischer ring
ABDOMEN :
Liver
Atypical or prolonged hepatitis
Hepatic cirrhosis
Hepatic coma
Hepatomegaly
Liver failure
High liver copper
Gastrointestinal
Esophageal varices
GENITOURINARY :
Kidneys
Renal tubular dysfunction
Renal calculi
SKELETAL :
Osteoporosis
Osteomalacia
Chondrocalcinosis
Limbs
Osteoarthritis
Joint hypermobility
NEUROLOGIC :
Central nervous system
Tremor
Dysarthria
Dysphagia
Personality changes
Dementia
Poor motor coordination
Dystonia
Drooling
Peripheral nervous system
Mixed demyelinating and axonal polyneuropathy (rare)
ENDOCRINE FEATURES :
Hypoparathyroidism
HEMATOLOGY :
Hemolytic anemia
LABORATORY ABNORMALITIES :
Low serum ceruloplasmin
High nonceruloplasmin-bound serum copper
High urinary copper
Proteinuria
Aminoaciduria
Glycosuria
Uricaciduria
Hyperphosphaturia
Hypercalciuria
MISCELLANEOUS :
Incidence in United States of 1 in 55,000
Incidence worldwide of 1 in 30,000 to 50,000
MENKES DISEASE(#309400)
Clinical Synopsis
INHERITANCE :
X-linked recessive
GROWTH :
Height
Short stature
Other
Intrauterine growth retardation
HEAD AND NECK :
Head
Microcephaly
Brachycephaly
Wormian bones
Face
Pudgy cheeks
CARDIOVASCULAR :
Vascular
Intracranial hemorrhage
SKELETAL :
Osteoporosis
Skull
Wormian bones
Limbs
Metaphyseal widening with spurs
SKIN, NAILS, HAIR :
Skin
Hypopigmentation
Hair
Steely, kinky, sparse hair
Twisted and partial breaks on magnification
NEUROLOGIC :
Central nervous system
Degenerative neurologic disorder with onset age 1 month
Hypertonia
Seizures
Intracranial hemorrhage
Hypothermia
LABORATORY ABNORMALITIES :
Low copper and ceruloplasmin
WILSON DISEASE (#277900)
Clinical Synopsis
INHERITANCE :
Autosomal recessive
HEAD AND NECK :
Eyes
Kayser-Fleischer ring
ABDOMEN :
Liver
Atypical or prolonged hepatitis
Hepatic cirrhosis
Hepatic coma
Hepatomegaly
Liver failure
High liver copper
Gastrointestinal
Esophageal varices
GENITOURINARY :
Kidneys
Renal tubular dysfunction
Renal calculi
SKELETAL :
Osteoporosis
Osteomalacia
Chondrocalcinosis
Limbs
Osteoarthritis
Joint hypermobility
NEUROLOGIC :
Central nervous system
Tremor
Dysarthria
Dysphagia
Personality changes
Dementia
Poor motor coordination
Dystonia
Drooling
Peripheral nervous system
Mixed demyelinating and axonal polyneuropathy (rare)
ENDOCRINE FEATURES :
Hypoparathyroidism
HEMATOLOGY :
Hemolytic anemia
LABORATORY ABNORMALITIES :
Low serum ceruloplasmin
High nonceruloplasmin-bound serum copper
High urinary copper
Proteinuria
Aminoaciduria
Glycosuria
Uricaciduria
Hyperphosphaturia
Hypercalciuria
MISCELLANEOUS :
Incidence in United States of 1 in 55,000
Incidence worldwide of 1 in 30,000 to 50,000
Abstract for presentation at 11th International Congress of Human Genetics
Dr Objoon Trachoo, Division of Medical Genetics and Molecular Medicine, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Pimjai Nipharak, Division of Hematology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Mrs Kanoknan Srichan, Division of Medical Genetics and Molecular Medicine, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Prasit Phowthongkum, Division of Medical Genetics, Department of Medicine, King Chulalongkorn Memorial Hospital, Chulalongkorn University, Bangkok, Thailand
Dr Suporn Chanjarunee, Division of Hematology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Surasak Kantachuvessiri, Division of Nephrology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Panus Chalermsaenyakorn, Department of Pathology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
A/Prof Thanyachai Sura, Division of Medical Genetics and Molecular Medicine, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
We report on Thai male identical twins who presented with hypochromic microcytic anemia, hepatosplenomegaly and gross hematuria since the age of 16. Hematological and hemoglobin analysis were consistent with a clinical diagnosis of beta thalassemia-Hb E disease and molecular diagnosis was confirmed by direct gene sequencing of all exons and splice junctions of beta globin gene. Neither structural renal disease nor renal stone was found from ultrasonographical findings. However, no specific cause of hematuria has been detected; therefore, renal biopsy was performed. Histological examination and specific immunological stain of renal tissue showed the evidence of IgA nephropathy. Recently, there were few reports of thalassemia disease associated with such particular renal involvement; thus far, we propose to include urinalysis screening in beta thalassemia-Hb E patients during follow-up in order to prevent further renal damage and end-stage renal disease.
Dr Pimjai Nipharak, Division of Hematology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Mrs Kanoknan Srichan, Division of Medical Genetics and Molecular Medicine, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Prasit Phowthongkum, Division of Medical Genetics, Department of Medicine, King Chulalongkorn Memorial Hospital, Chulalongkorn University, Bangkok, Thailand
Dr Suporn Chanjarunee, Division of Hematology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Surasak Kantachuvessiri, Division of Nephrology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Dr Panus Chalermsaenyakorn, Department of Pathology, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
A/Prof Thanyachai Sura, Division of Medical Genetics and Molecular Medicine, Department of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
We report on Thai male identical twins who presented with hypochromic microcytic anemia, hepatosplenomegaly and gross hematuria since the age of 16. Hematological and hemoglobin analysis were consistent with a clinical diagnosis of beta thalassemia-Hb E disease and molecular diagnosis was confirmed by direct gene sequencing of all exons and splice junctions of beta globin gene. Neither structural renal disease nor renal stone was found from ultrasonographical findings. However, no specific cause of hematuria has been detected; therefore, renal biopsy was performed. Histological examination and specific immunological stain of renal tissue showed the evidence of IgA nephropathy. Recently, there were few reports of thalassemia disease associated with such particular renal involvement; thus far, we propose to include urinalysis screening in beta thalassemia-Hb E patients during follow-up in order to prevent further renal damage and end-stage renal disease.
Thalassemia is a genetic disorders that are characterized with chronic anemia. The disease is cause from the gene that control hemoglobin production. Hemoglobins are proteins that are the major components of red blood cell. Human carries mutation of thalassemia genes may be asymptomatic or have mild anmia, or severe until they died before birth.
In Thailand, a south east Asian country, there are more than 30 percent having one mutated hemoglobin genes. We call this group as carrier. They will be healthy and asymptomatic, but they can inherited their mutated gene to their offsprings. If their couples are also the same type carriers, their off springs have chance to have mutated genes both from thier parents that will produce diseases. The number of thalassemia patients in Thailad is about 600000.
And each year, couples at risk (both are carriers) give thalassemic baby for more than 10000 annually.
Ther are two common type of thalassemia in Thailand: alpha-thalassemia and beta-thalassemia.
In general, everyone are inherited the genetic materials half from their father and the rest from their mother. Thalassemia is a disease transfered in recessive fashion, which means that you must have mutated genes from both parents to have disease. Besides, the mutation must be on the same gene. For example, alpha-thalassemia gene mutation should pair with alpha-thalassemia gene mutation to produce alpha-thalassemia patients. Who have one mutated gene of beta-type and onother mutated gene of alpha-type are healthy.
Thalassemia is the result of decreasing production of hemoglobin leading to unbalanced between each type of hemoglobin. In normal, two chain of alpha hemoglobin will be paired with two chains of beta hemoglobin. In the non-balancing situation, the excess hemoglobin will be aggregated and cause changing of red cell shape and vulnerability. red cell will be survived less, be easily destroy. These all lead to chronic anemia. Organ that produce red blood cell i.e. bone marrow, liver, and spleen have to be expanded for increasing production rate, so we will see these patients have a big and tall skull, fragile bone, big liver, big spleen. In severe cases, this occured early in thier life, and inevitable cause of death in their toddler. Milder case will be survived to their teen with regular blood transfusion. The drawbcak of blood transfusion is the iron stored in the blood cell will be accumulated in patient's heart, liver, skin, pancreas, and so on lead to malfunction of that organs that are fatal if lefted untreated. Nowadays, in the most severe type that will be dead before birth, we have technology to test the fetus and termination before birth or have complication will be done if test positive. In severe birth infants, we can treated with bone marrow transplantation, but this is a very expensive methods and there is a high death rate from this procedure and not everyone can performed these due to unmatched tissues. Milder case will receive regular blood transfusion to prevent abnormal growth and bone change, and iron-chelator will be given to prevent long term complication for iron-excess. Prevention is more appropriate practice, and prenatal screening method is the one of most success genetic project in our country. In the future , next four or five generation, we will have less thalassemia patients.
In Thailand, a south east Asian country, there are more than 30 percent having one mutated hemoglobin genes. We call this group as carrier. They will be healthy and asymptomatic, but they can inherited their mutated gene to their offsprings. If their couples are also the same type carriers, their off springs have chance to have mutated genes both from thier parents that will produce diseases. The number of thalassemia patients in Thailad is about 600000.
And each year, couples at risk (both are carriers) give thalassemic baby for more than 10000 annually.
Ther are two common type of thalassemia in Thailand: alpha-thalassemia and beta-thalassemia.
In general, everyone are inherited the genetic materials half from their father and the rest from their mother. Thalassemia is a disease transfered in recessive fashion, which means that you must have mutated genes from both parents to have disease. Besides, the mutation must be on the same gene. For example, alpha-thalassemia gene mutation should pair with alpha-thalassemia gene mutation to produce alpha-thalassemia patients. Who have one mutated gene of beta-type and onother mutated gene of alpha-type are healthy.
Thalassemia is the result of decreasing production of hemoglobin leading to unbalanced between each type of hemoglobin. In normal, two chain of alpha hemoglobin will be paired with two chains of beta hemoglobin. In the non-balancing situation, the excess hemoglobin will be aggregated and cause changing of red cell shape and vulnerability. red cell will be survived less, be easily destroy. These all lead to chronic anemia. Organ that produce red blood cell i.e. bone marrow, liver, and spleen have to be expanded for increasing production rate, so we will see these patients have a big and tall skull, fragile bone, big liver, big spleen. In severe cases, this occured early in thier life, and inevitable cause of death in their toddler. Milder case will be survived to their teen with regular blood transfusion. The drawbcak of blood transfusion is the iron stored in the blood cell will be accumulated in patient's heart, liver, skin, pancreas, and so on lead to malfunction of that organs that are fatal if lefted untreated. Nowadays, in the most severe type that will be dead before birth, we have technology to test the fetus and termination before birth or have complication will be done if test positive. In severe birth infants, we can treated with bone marrow transplantation, but this is a very expensive methods and there is a high death rate from this procedure and not everyone can performed these due to unmatched tissues. Milder case will receive regular blood transfusion to prevent abnormal growth and bone change, and iron-chelator will be given to prevent long term complication for iron-excess. Prevention is more appropriate practice, and prenatal screening method is the one of most success genetic project in our country. In the future , next four or five generation, we will have less thalassemia patients.
Common situation in genetic counseling
Today, a medical student asked me about her relative was pregnant and she found that her baby had thalassemia from the prenatal screening. Her doctor gave her a chance to terminate the pregnancy. This student shown me the genotype: Hb Malay (Mutation at codon 19)/ Hb E. This is called compound heterozygotes, in contrary to double hetrozygotes that have mutation on different genes. To answer this question, we should know about the severity of phenotypes, effect to mother.Fortunately, thalassemia is one of the most common and the most throughly study genetic diseases, so we have some information to cope with that. But genotype-phenotype correlation is not an easy issue, there are many factors beyond the mutation itself to correctly predict that. Like these situations, Hb Maly/Hb E infants are not severe (thalassemia intermedia), but the characteristics are widely range from asymptomatic, need some blood transfusion or more frequent need of blood transfusion until required iron chelating therapy or splenectomy in mid-life. There is no specific curative treatment for this group, while beta thallassemia major is so severe and will be died in first two or three years of life, so high mortality-procedure dependent like bone marrow transplantation is accepted.
So the best recommendation for this situation is give all the informations needed to make a good decision for those counselee i.e. severity of baby, effect for pregnancy, treatment plan for baby, side effect of termination, choices, etc. And let the families making their own decision because they have to know what is the best way for their future life. Don't use our standard, believes, social issues to force or coercion them.
So the best recommendation for this situation is give all the informations needed to make a good decision for those counselee i.e. severity of baby, effect for pregnancy, treatment plan for baby, side effect of termination, choices, etc. And let the families making their own decision because they have to know what is the best way for their future life. Don't use our standard, believes, social issues to force or coercion them.
How to be up-to-date in genetics?
1. follow the most reliable genetic sites
2. follow the best genetic blogs
3. Use Rss web feed and follow the genetics journal
4. Use service/tools eg. uptodate
5. follow the blog carnivals
6. follow the genetic wiki
7. your choice
Fromhttp://scienceroll.com/medicine-20/
That sound pretty good, but keep in mind the accuracy, the reliablity of the sources. Every thing can be found on iternet without any prove.
2. follow the best genetic blogs
3. Use Rss web feed and follow the genetics journal
4. Use service/tools eg. uptodate
5. follow the blog carnivals
6. follow the genetic wiki
7. your choice
Fromhttp://scienceroll.com/medicine-20/
That sound pretty good, but keep in mind the accuracy, the reliablity of the sources. Every thing can be found on iternet without any prove.
Subscribe to:
Posts (Atom)