DNA preparation for cloning from mRNA
1.see for abundance of mRNA -partial answer about function
eg> young RBC - large amount of Hemoglobin mRNA, chicken fallopian tube - ovalbumin mRNA 100000 molecule/cell( in contrast others species (12507) toally less than 100000
2.do not need post transcriptional modification machine of eukaryotes
Method
1. mRNA--> reverse transcriptase --> cDNA (complementary DNA)
Primers: oligo dT --> digest RNA with alkaline
sscDNA have a hook use as primer for DNA polymerase --> ds cDNA with hair pin end
S1 nuclease--> two blunt end
primer for this second strand is not so effective and S1 nuclease can shorten or unequal cut
2. use RNAse h replace alkaline --> random digest so RNA can be use as primer
second strand production by DNA polymerase I--> T4 DNA polymerase cut to be blunt end
DNA preparation by chemical synthesis
up to 50 nt
oligonucleotides and link with DNA ligase
eg: interferon gene : 66 oligonucleotides to 514 bp
commonly use for probe, primer, linker synthesis
Method
1. Phosphate triester
add protective group at amino group of A, C (benzoyl), G (isobutyryl)
add protective roup at 5' with dimethoxytrityl chloride (CH3O)2Tr-
add p-chlorophenylphosphorodichloride at 3' to link with another nucleotide [with 3' protected (berta cyanoethanol) and dimethoxytrityl at 5' removal with benzebnesulfonic]
react with triisopropylbenzenesulfonyl chloride
--> all protected dinuleotide--> select removal to control direction of synthesis
can be automated when attached with solid phase
10-20 nt in 2-3 days
2. Phosphite triester
linker is nucleoside 3- phosphoramidite
different protected group and removers
15 min 50 bp good quality
Monday, 26 November 2007
genetic engineering (10)DNA preparation for cloning
DNa preparation from cell
keep Easy, Feasible, Simple
Animal cells:
Liver, spleen,kidney, blood, cell culture
Liver: fast 24 hr to decrease glycogen
Spleen: good source, large amount
fresh tissue or frozen tissue (-80 can keep for 1 year)
Principles see isolation DNA protocol
1. Cell lysis by detergent: SDS, sarkosyl, proteinase K
2. Extract protein and cell debris with phenol
3. Pecipitate DNA with ethanol, isopropanol
3. Purification by high g centrifugation in CsCl, ethidium bromide
Electrophoresis to measure size , quantitate with OD method
Plant cells : ask botanist I don't know much about these.
keep Easy, Feasible, Simple
Animal cells:
Liver, spleen,kidney, blood, cell culture
Liver: fast 24 hr to decrease glycogen
Spleen: good source, large amount
fresh tissue or frozen tissue (-80 can keep for 1 year)
Principles see isolation DNA protocol
1. Cell lysis by detergent: SDS, sarkosyl, proteinase K
2. Extract protein and cell debris with phenol
3. Pecipitate DNA with ethanol, isopropanol
3. Purification by high g centrifugation in CsCl, ethidium bromide
Electrophoresis to measure size , quantitate with OD method
Plant cells : ask botanist I don't know much about these.
genetic engineering (9)
interest DNA: genomic DNA, complementary DNA chemical or enzymatic synthetic
+vector : plasmid,phage, cosmid, phagemid, BAC, YAC
=recombinant DNA-->host: bacteria, yeast, fungi, animal cells, plant cells
+vector : plasmid,phage, cosmid, phagemid, BAC, YAC
=recombinant DNA-->host: bacteria, yeast, fungi, animal cells, plant cells
genetic engineering (8)
DNA modifying enzymes
T4 polynucleotide kinase-T4 Ecoli ATP at 5' or exchange reaction-5' labeling*****
Alkaline phosphatase-bacterial AP(BAP-heat satble) or calf intestinal AP (CIP-heat labile)-5' digest Phosphate group ,prevent ligase action
DNA ligase-E coli ligase (NAD) or T4DNA ligase (ATP) nick repair or link cohesive end or blunt end (T4 ligase)
Terminal deoxynucleotidyl transferase-bovine thymus-3' add dNTP without template add complementary sequence at one end of vector and one end of gene
Nuclease
NUclease Bal 31-Alteromonas espejiana Bal 31 5'->3' exonuclease , ssDNA endonuclease
Exonuclease III-E coli3'->5' exonuclease (cannot use 3' protruding)
S1 Nuclease-Aspergillus oryzae-endonuclease ssDNA, ssRNA, nick or gap in duplex DNA, or RNA use to detect non-complete complementary, produce blut end, hairpin cDNA break
Mung beab nuclease (MB nuclease)-Mung bean-ssDNA, ssRNA, gap in duplex DNA, or RNA
DNAse I- cow pancreas- endonuclease ss,ds, with Mg - random, with Mn-blunt end or 1-2 bp protruding end
RNase -E coli digest RNA hybrid with DNA
T4 polynucleotide kinase-T4 Ecoli ATP at 5' or exchange reaction-5' labeling*****
Alkaline phosphatase-bacterial AP(BAP-heat satble) or calf intestinal AP (CIP-heat labile)-5' digest Phosphate group ,prevent ligase action
DNA ligase-E coli ligase (NAD) or T4DNA ligase (ATP) nick repair or link cohesive end or blunt end (T4 ligase)
Terminal deoxynucleotidyl transferase-bovine thymus-3' add dNTP without template add complementary sequence at one end of vector and one end of gene
Nuclease
NUclease Bal 31-Alteromonas espejiana Bal 31 5'->3' exonuclease , ssDNA endonuclease
Exonuclease III-E coli3'->5' exonuclease (cannot use 3' protruding)
S1 Nuclease-Aspergillus oryzae-endonuclease ssDNA, ssRNA, nick or gap in duplex DNA, or RNA use to detect non-complete complementary, produce blut end, hairpin cDNA break
Mung beab nuclease (MB nuclease)-Mung bean-ssDNA, ssRNA, gap in duplex DNA, or RNA
DNAse I- cow pancreas- endonuclease ss,ds, with Mg - random, with Mn-blunt end or 1-2 bp protruding end
RNase -E coli digest RNA hybrid with DNA
Genetic engineering (7)
Polymerase and exonuclease
E coli DNA polymerase I
1 chain polypeptide
109000 Dalton
5'->3' polymerase
5'->3" exonuclease
3'-> 5" exonuclease
proof reading and repair DNA
nick translation
DNA labeling- low concentration of DNAse, add label
Klenow fragment,large fragment
1 chain polypeptide
Pol I--> typsin, subtilisin 76000 Dalton
C-terminal, no 5'->3' exonuclease
use for DNA synthesis from RNA, enzymatic sequencing, add nucleotide for 5 protruding end, replacement or exchange reaction of 3' blut end
T4 DNA polymerase
same as Klenow activity, same use
but 3'->5' activity 200X
dNTP concentration control polymerase activity
RNA dependent DNA polymerase, reverse transcriptase
RNA virus enzyme
5'->3' DNA synthesis from RNA template
need ssRNA, or ssDNA as template and primer
make cDNA from mRNA, 3' fill in
Polymerase Chain Reaction (PCR)
E coli DNA polymerase I
1 chain polypeptide
109000 Dalton
5'->3' polymerase
5'->3" exonuclease
3'-> 5" exonuclease
proof reading and repair DNA
nick translation
DNA labeling- low concentration of DNAse, add label
Klenow fragment,large fragment
1 chain polypeptide
Pol I--> typsin, subtilisin 76000 Dalton
C-terminal, no 5'->3' exonuclease
use for DNA synthesis from RNA, enzymatic sequencing, add nucleotide for 5 protruding end, replacement or exchange reaction of 3' blut end
T4 DNA polymerase
same as Klenow activity, same use
but 3'->5' activity 200X
dNTP concentration control polymerase activity
RNA dependent DNA polymerase, reverse transcriptase
RNA virus enzyme
5'->3' DNA synthesis from RNA template
need ssRNA, or ssDNA as template and primer
make cDNA from mRNA, 3' fill in
Polymerase Chain Reaction (PCR)
- Template with known at least head or end sequence
- Design small oligonucletide complement with 3' of each end (20-35 b)
- Mix large amount of primers with template DNA
- Deanneal template with heat
- Reanneal of primer to template
- DNA polymerase will extend DNA 5'->3'
- double each round
Taq DNA polymerase - heat stable
usually temperature setting
deanneal-95 1 min
anneal-60 1 min
extension-72 1 min
30-40 rounds
genetic engineering (6)
Enzymes for cloning
1. restriction enzyme or restriction endonulease
defense mechanisms of bacteria: restriction system, modification system (methylation)
Type
Type 2 is the enzyme use in genetic engineering
single polypeptide
cleave site in or near restriction recognition
need only Magnesium ion
restriction only
Type 1
3 polypeptides
DNAse, methyla
specific recognition site, but not the cleave site (far 400-7000 bp)
need Mg, ATP, SAM
no function after nuclease
Type 3
2 polypeptides
DNAse, methylase
cleave about 25-27 b from RS
need Mg, ATP
Type 2 naming Italic font
First letter -capital letter, genus
SEcond and third-small letter, species
strain-optional
Roman number-from discovery
RS - 4-6 bp -axis of symmetry, palindrome(not necessary)
sticky or cohesive end
5' protruding or 3" protruding
blunt or flush end
isoschizomer-same RS, not necessary same cleave site
90 RS
probabilties to find RS 4 bp= 25 bp
probabilities to find RS 6 bp= 4096 bp
conditions:
Tris HCl
Mg
NaCl
2-mercaptoethanol
pH 7.2-7.6
37 degree celcius
restrict to commercial recommendation to avoid star activity!
enzyme usually in glycerol so avoid too much enzyme
restriction map
-restriction enzymes
-DNA gel electrophoresis
polyacrylamide- 6 bp (20% acrylamide)-1000 bp (3% acrylamide)
agarose-70 bp (3%)-80000 bp (0.1%)
-migrate inverse log of bp
-visulaize-autoradiograph, ethidium bromide
Method 1- partial digestion of 1 enzyme
Method 2-complete digestion of more than 1 enzyme
Southern blot
-transfer to membrane (nitocellulose, nylon)
-denature to siigle stranded
-hybridization with DNA probe-autoradiograph
Northern blot- RNA
Western blot- protein
mutation can create or delete RS lead to change of restrction pattern-restriction fragment length polymorphism (RFLP)
1. restriction enzyme or restriction endonulease
defense mechanisms of bacteria: restriction system, modification system (methylation)
Type
Type 2 is the enzyme use in genetic engineering
single polypeptide
cleave site in or near restriction recognition
need only Magnesium ion
restriction only
Type 1
3 polypeptides
DNAse, methyla
specific recognition site, but not the cleave site (far 400-7000 bp)
need Mg, ATP, SAM
no function after nuclease
Type 3
2 polypeptides
DNAse, methylase
cleave about 25-27 b from RS
need Mg, ATP
Type 2 naming Italic font
First letter -capital letter, genus
SEcond and third-small letter, species
strain-optional
Roman number-from discovery
RS - 4-6 bp -axis of symmetry, palindrome(not necessary)
sticky or cohesive end
5' protruding or 3" protruding
blunt or flush end
isoschizomer-same RS, not necessary same cleave site
90 RS
probabilties to find RS 4 bp= 25 bp
probabilities to find RS 6 bp= 4096 bp
conditions:
Tris HCl
Mg
NaCl
2-mercaptoethanol
pH 7.2-7.6
37 degree celcius
restrict to commercial recommendation to avoid star activity!
enzyme usually in glycerol so avoid too much enzyme
restriction map
-restriction enzymes
-DNA gel electrophoresis
polyacrylamide- 6 bp (20% acrylamide)-1000 bp (3% acrylamide)
agarose-70 bp (3%)-80000 bp (0.1%)
-migrate inverse log of bp
-visulaize-autoradiograph, ethidium bromide
Method 1- partial digestion of 1 enzyme
Method 2-complete digestion of more than 1 enzyme
Southern blot
-transfer to membrane (nitocellulose, nylon)
-denature to siigle stranded
-hybridization with DNA probe-autoradiograph
Northern blot- RNA
Western blot- protein
mutation can create or delete RS lead to change of restrction pattern-restriction fragment length polymorphism (RFLP)
Sunday, 25 November 2007
genetic engineering (5)
RA synthesis
5'->3' direction
no primer
DNA dependent RNA polymerase (RNA polymerase)
ATP, GTP, CTP, UTP
anticoding strand, template strand
promotor -RNA polymerase (alpha, beta, delta)binding site - 5' ustream to transcription initiation site (+1)
termination -Rho protein, inverted repeat
prokaryote - -35 and - 10 consensus sequence- Pribnow box
Eukaryote -75 CAAT box -5'GGNCAATCT3', -25 TATA box,Hogness box - 5' TATAAA3'
higher organisms
RNA pol 1 polA-rRNA except 5 SrRNA, super repeat promotor
RNA pol2 polB-mRNA- normal promotor, AAUAAA-polyadenylation signal (20-30 b before 3')
RNA pol3 polC-tRNA, 5srRNA, scRNA, internal control region (ICR)
Posttranscriptional modification
tRNA- methylation, oxygen-->sulfur
rRNA-methylation, shortening
mRNA
-7 methyl guanosine capping
-poly Adenylation tail
-splicing -exclude intron-snRNPs, some self splicing
alpha interferon - no intron
beta globin -2 intron
factor VIII-25 intron
human thyroglobulin- > 40 intron
5'->3' direction
no primer
DNA dependent RNA polymerase (RNA polymerase)
ATP, GTP, CTP, UTP
anticoding strand, template strand
promotor -RNA polymerase (alpha, beta, delta)binding site - 5' ustream to transcription initiation site (+1)
termination -Rho protein, inverted repeat
prokaryote - -35 and - 10 consensus sequence- Pribnow box
Eukaryote -75 CAAT box -5'GGNCAATCT3', -25 TATA box,Hogness box - 5' TATAAA3'
higher organisms
RNA pol 1 polA-rRNA except 5 SrRNA, super repeat promotor
RNA pol2 polB-mRNA- normal promotor, AAUAAA-polyadenylation signal (20-30 b before 3')
RNA pol3 polC-tRNA, 5srRNA, scRNA, internal control region (ICR)
Posttranscriptional modification
tRNA- methylation, oxygen-->sulfur
rRNA-methylation, shortening
mRNA
-7 methyl guanosine capping
-poly Adenylation tail
-splicing -exclude intron-snRNPs, some self splicing
alpha interferon - no intron
beta globin -2 intron
factor VIII-25 intron
human thyroglobulin- > 40 intron
genetic engineering (4)
DNA replication
semiconservative
1.helix-destabilizing protein-helicase
2.single strand DNA binding protein (SBP, DBP)
3. DNA gyrase or topoisomerase-replication fork
4.primase-synthesize small RNA primer
5.DNA polymerase
E coli- pol3 leading strand 5'-->3', lagging strand small fragment (Ogazaki fragment), pol I 5'->3' exonuclease)
polI (C-terminal -polymerase, 3'-5' exonuclease -Klenov fragment, short N-terminal, 5'-3' exonuclease) -proofreading
Mammals-alpha-nuclear-polymerase, beta-nuclear-repair, delta -nuclear similar alpha, gamma-mitochondria-mitochondrial DNAreplication
6. DNA ligase
bidirectional
E coli- 1 replicon
Yeast-500 replicon
Drosophila-3500 replicon
Toad-15000 replicon
Mice 25000 replicon
plant -35000 replicon
Model
1. Theta model-circular DNA-bacteria, mitochondria
2. Sigma model-rolling circle model: phage lambda circular dsDNA-nick, unwheel, produce linear DNA-concatamer, catenated DNA, phage M13 circular ssDNA --> circular dsDNA--> linear ssDNA-->circular ssDNA
3. Y-shaped model-phage T7 linear dsDNA, higher organisms- chromosome
telomerase - terminal deoxynucleotidyltransferase - repeat CCCTAA---> inverted to complementary
found in egg and sperm only
so shorten of chromosome each replication 50-60 times --.senescence
semiconservative
1.helix-destabilizing protein-helicase
2.single strand DNA binding protein (SBP, DBP)
3. DNA gyrase or topoisomerase-replication fork
4.primase-synthesize small RNA primer
5.DNA polymerase
E coli- pol3 leading strand 5'-->3', lagging strand small fragment (Ogazaki fragment), pol I 5'->3' exonuclease)
polI (C-terminal -polymerase, 3'-5' exonuclease -Klenov fragment, short N-terminal, 5'-3' exonuclease) -proofreading
Mammals-alpha-nuclear-polymerase, beta-nuclear-repair, delta -nuclear similar alpha, gamma-mitochondria-mitochondrial DNAreplication
6. DNA ligase
bidirectional
E coli- 1 replicon
Yeast-500 replicon
Drosophila-3500 replicon
Toad-15000 replicon
Mice 25000 replicon
plant -35000 replicon
Model
1. Theta model-circular DNA-bacteria, mitochondria
2. Sigma model-rolling circle model: phage lambda circular dsDNA-nick, unwheel, produce linear DNA-concatamer, catenated DNA, phage M13 circular ssDNA --> circular dsDNA--> linear ssDNA-->circular ssDNA
3. Y-shaped model-phage T7 linear dsDNA, higher organisms- chromosome
telomerase - terminal deoxynucleotidyltransferase - repeat CCCTAA---> inverted to complementary
found in egg and sperm only
so shorten of chromosome each replication 50-60 times --.senescence
Genetic engineering (3)
Structure and type of RNA
RNA - 5X DNA
transfer RNA (tRNA)80-100 type
5S ribosomal RNA 1-2 type
5.8 Sribosomal RNA 1 type
16 S ribosomal RNA 1 type
23 S ribosomal RNA 1 type
18 S ribosomal RNA 1 type
28 S ribosomal RNA 1 type
messenger RNA (mRNA) many thousands type
heterogeneous nuclear RNA (hnRNA) many thousands type
small cytoplasmic RNA (scRNA)
small nuclear RNA (snRNA)
Size, organization, complexity of Genomes
Size and organization of genomes
1000 bp to 10000 mbp
dsDNA except virus: ssDNA, dsDNA, ssRNA, dsRNA, linear or circular, single or multiple
bacteria-circular dsDNA+protein
eukaryote-linear dsDNA (multile)+histone, non histone protein
left hand super helix 1.8 rounds, 145 bpDNA(2nm)+2H2A, 2H2B, 2H3, 2H4- nucleosome core particle(10nm)--> solenoid(30nm)-->filament(300nm)-->supercoiled(700nm)-->chromosome (metaphase 1400nm)
60 bp linker DNA+H1
heterochromatin-inactive-centromere, telomere
active chromatin-10%
packing ratio=DNA length/Final length average 1000-2000
Complexity of genome
unique sequence, repetitive sequence
slow component 1 copies-45% of genome
intermediate component 350 copies-30% of genome
fast component 500000 copies-25% of genome
satellite, minisatellite, microsatellite
Important of DNA
DNA replication
DNA transcription
Complexity of genome
RNA - 5X DNA
transfer RNA (tRNA)80-100 type
5S ribosomal RNA 1-2 type
5.8 Sribosomal RNA 1 type
16 S ribosomal RNA 1 type
23 S ribosomal RNA 1 type
18 S ribosomal RNA 1 type
28 S ribosomal RNA 1 type
messenger RNA (mRNA) many thousands type
heterogeneous nuclear RNA (hnRNA) many thousands type
small cytoplasmic RNA (scRNA)
small nuclear RNA (snRNA)
Size, organization, complexity of Genomes
Size and organization of genomes
1000 bp to 10000 mbp
dsDNA except virus: ssDNA, dsDNA, ssRNA, dsRNA, linear or circular, single or multiple
bacteria-circular dsDNA+protein
eukaryote-linear dsDNA (multile)+histone, non histone protein
left hand super helix 1.8 rounds, 145 bpDNA(2nm)+2H2A, 2H2B, 2H3, 2H4- nucleosome core particle(10nm)--> solenoid(30nm)-->filament(300nm)-->supercoiled(700nm)-->chromosome (metaphase 1400nm)
60 bp linker DNA+H1
heterochromatin-inactive-centromere, telomere
active chromatin-10%
packing ratio=DNA length/Final length average 1000-2000
Complexity of genome
unique sequence, repetitive sequence
slow component 1 copies-45% of genome
intermediate component 350 copies-30% of genome
fast component 500000 copies-25% of genome
satellite, minisatellite, microsatellite
Important of DNA
DNA replication
DNA transcription
Complexity of genome
Genetic engineering (2)
Structure and function of Deoxyribonucleic Acid (DNA)
Structure and behavior of DNA and RNA (Ribonucleic acid)
Chemical composition
Nucleotide=Base+Sugar+Phosphate group
Nucleotide=(Purine: adenine(A), guanine(G) or Pyrimidine: Cytosine(C), Thymidine (T-DNA only), uracil (U-RNA only)+ Deoxyribose (DNA) or Ribose(RNA)
5' --> 3 '
Structure of DNA
B-DNA
double helix
antiparallel
base -hydrogen bone- base
A=T, G-triple bond-C in horizontal plane
sugar back bone
phosphate outside
3.4 nm height for each round 10 base
diameter 2 nm
A-DNA
relative humidity 75%, Sodium, Potassium, or Cesium
base diagonal 20 degree, each base differ 0.26 nm
11-12 bp each round
diameter 2.3 nm
antiparallel
RNA, or DNA-RNA hybrid
Z-DNA
poly dGC,polydAC
each base differ 0.37 nm
12 bp per each round
diameter 1.8 nm
Zigzag-like
stabilized with replace C in poly dGC with 5-methyl C
Denature (melting)and renature(association, annealing)
melting temperature (Tm) -temperature that have equal ssDNA and dsDNA
pHm-pH that have equal ssDNA and dsDNA
urea and formamide-decrease Tm
monovalent or divalent cation concentration-increase Tm
G+C/A+T increase Tm. pHm
GC increase 1%, Tm increase 0.4 degree
renaturation between different species of nucleic acid-hybridization: solution, filter
Other qualities of DNA
1. UV absorption: absorbance (A) or optical density (OD) peak at 260
OD of 1 mg/ml of DNA, RNAA, oligonucleotide = 20, 25, 30
OD increase with temperature (ssDNA-> increase OD)
2. Acid-base:stable at pH 4-5, depurination at pH <3, base donot react with DNA, but destroy phosphodiester bond of RNA (react at OH)
3.sedimentation
velocity sedimentation: generate gradient column of sucrose or potassium citrate-add DNA mixture-centrifuge-highMW down, low MW up
equilibrium sedimentation:CS-->spin down centrifuge-->diffusion until equilibrium-add Mixture buoncy of DNA=Cs, protein-top, RNA-bottom
Structure and behavior of DNA and RNA (Ribonucleic acid)
Chemical composition
Nucleotide=Base+Sugar+Phosphate group
Nucleotide=(Purine: adenine(A), guanine(G) or Pyrimidine: Cytosine(C), Thymidine (T-DNA only), uracil (U-RNA only)+ Deoxyribose (DNA) or Ribose(RNA)
5' --> 3 '
Structure of DNA
B-DNA
double helix
antiparallel
base -hydrogen bone- base
A=T, G-triple bond-C in horizontal plane
sugar back bone
phosphate outside
3.4 nm height for each round 10 base
diameter 2 nm
A-DNA
relative humidity 75%, Sodium, Potassium, or Cesium
base diagonal 20 degree, each base differ 0.26 nm
11-12 bp each round
diameter 2.3 nm
antiparallel
RNA, or DNA-RNA hybrid
Z-DNA
poly dGC,polydAC
each base differ 0.37 nm
12 bp per each round
diameter 1.8 nm
Zigzag-like
stabilized with replace C in poly dGC with 5-methyl C
Denature (melting)and renature(association, annealing)
melting temperature (Tm) -temperature that have equal ssDNA and dsDNA
pHm-pH that have equal ssDNA and dsDNA
urea and formamide-decrease Tm
monovalent or divalent cation concentration-increase Tm
G+C/A+T increase Tm. pHm
GC increase 1%, Tm increase 0.4 degree
renaturation between different species of nucleic acid-hybridization: solution, filter
Other qualities of DNA
1. UV absorption: absorbance (A) or optical density (OD) peak at 260
OD of 1 mg/ml of DNA, RNAA, oligonucleotide = 20, 25, 30
OD increase with temperature (ssDNA-> increase OD)
2. Acid-base:stable at pH 4-5, depurination at pH <3, base donot react with DNA, but destroy phosphodiester bond of RNA (react at OH)
3.sedimentation
velocity sedimentation: generate gradient column of sucrose or potassium citrate-add DNA mixture-centrifuge-highMW down, low MW up
equilibrium sedimentation:CS-->spin down centrifuge-->diffusion until equilibrium-add Mixture buoncy of DNA=Cs, protein-top, RNA-bottom
Genetic engineering (1)
Introduction
Basic genetics to genetic engineering
1866 Mendel - Principle of Inheritance
1900 de Vries, Correns, van Tschermak - rediscover Mendel's rule
1905 Bateson - word "Genetics"
1901 Johannsen - word "Genes"
1928 Avery, Macleod, McCarthy- DNA is genetic material
1953 Watson, Crick - double helix DNA
Beginning of genetic engineering techniques
1970 Nathans, Smith - restriction enzymes
1970 reverse transcriptase
1976 antibody diversities
1977 split genes: introns and exons
1977 Sequencing techniques
1982 Supermice: Growth Hormone Gene Transfer
1983 Ribozymes
1985 Polymerase chain reaction (PCR), DNA fingerprint
1990 CFTR gene for cystic fibrosis
1993 IT15 gene for Huntington disease
1997 Telomerase gene
2003 First draft of Human Genome
Basic genetics to genetic engineering
1866 Mendel - Principle of Inheritance
1900 de Vries, Correns, van Tschermak - rediscover Mendel's rule
1905 Bateson - word "Genetics"
1901 Johannsen - word "Genes"
1928 Avery, Macleod, McCarthy- DNA is genetic material
1953 Watson, Crick - double helix DNA
Beginning of genetic engineering techniques
1970 Nathans, Smith - restriction enzymes
1970 reverse transcriptase
1976 antibody diversities
1977 split genes: introns and exons
1977 Sequencing techniques
1982 Supermice: Growth Hormone Gene Transfer
1983 Ribozymes
1985 Polymerase chain reaction (PCR), DNA fingerprint
1990 CFTR gene for cystic fibrosis
1993 IT15 gene for Huntington disease
1997 Telomerase gene
2003 First draft of Human Genome
Thursday, 22 November 2007
Treament of genetic diseases in the real world 9: familial hypokalemic periodic paralysis
What is hypokalemic periodic paralysis?
Hypokalemic periodic paralysis is a condition that causes episodes of extreme muscle weakness typically beginning in childhood or adolescence. Most often, these episodes involve a temporary inability to move muscles in the arms and legs. Attacks cause severe weakness or paralysis that usually lasts from hours to days. Some people may have episodes almost every day, while others experience them weekly, monthly, or only rarely. Attacks can occur without warning or can be triggered by factors such as rest after exercise, a viral illness, or certain medications. Often, a large, carbohydrate-rich meal or vigorous exercise in the evening can trigger an attack upon waking the following morning. Although affected individuals usually regain their muscle strength between attacks, repeated episodes can lead to persistent muscle weakness later in life.
People with hypokalemic periodic paralysis have reduced levels of potassium in their blood (hypokalemia) during episodes of muscle weakness. Researchers are investigating how low potassium levels may be related to the muscle abnormalities in this condition.
How common is hypokalemic periodic paralysis?
Although its exact prevalence is unknown, hypokalemic periodic paralysis is estimated to affect 1 in 100,000 people. Men tend to experience symptoms of this condition more often than women.
Diagnosis/testing. The diagnosis of HOKPP is based on a history of episodes of flaccid paralysis; low serum concentration of potassium (<0.9 to 3.0 mmol/L) during attacks; the absence of myotonia clinically and on electromyography (EMG) (with the exception of one family with heat-induced myotonia and cold-induced HOKPP); and a family history consistent with autosomal dominant inheritance. Molecular genetic testing identifies disease-causing mutations in CACNA1S or SCN4A in 80% of individuals meeting clinical diagnostic criteria. Of all individuals with HOKPP, about 55-70% have mutations in CACNA1S and about 8-10% in SCN4A. Such testing is clinically available.
Management. Treatment of a paralytic crisis by administration of potassium by mouth or IV aims to normalize the serum concentration of potassium and to shorten the paralytic episode. ECG and blood potassium concentration must be monitored during treatment. Surveillance depends on the affected individual's symptoms and response to preventive treatment. Neurologic examination should focus on muscle strength in the legs to detect permanent weakness associated with myopathy. A number of factors can trigger paralytic attacks: unusually strenuous effort, excess of carbohydrate-rich meals, sweets, and alcohol should be avoided; oral or intravenous corticosteroids should be used with care; glucose infusion should be replaced by another type of infusion.
Potassium in doses of 0.2 to 0.4 mmol/kg is administered orally every 15 to 30 minutes over one to three hours.
If the individual is unable to swallow or does not tolerate potassium by mouth, potassium may be administered intravenously. In that case, it must be diluted in 5% mannitol rather than in glucose or sodium chloride, which trigger crises in individuals with HOKPP. The concentration of potassium administered intravenously must not exceed 40 mmol/L and the flow must not exceed 20 mmol/hour or 200-250 mmol/day; administration must be stopped when the serum potassium concentration is normalized, even if weakness persists.
Because the hypokalemia and subsequent changes in potassium
Genetic counseling. HOKPP is inherited in an autosomal dominant manner. Most individuals diagnosed with HOKPP have an affected parent. The proportion of cases caused by a de novo gene mutation is unknown. Offspring of a proband have a 50% risk of inheriting the mutation. Penetrance is about 90% in males and may be as low as 50% in females depending on the causative mutation. Prenatal testing is possible if the disease-causing mutation has been identified in the proband; however, requests for prenatal testing for conditions such as HOKPP that do not affect intellect and have some treatment available are not common.
POTASSIUM
Hypokalemic periodic paralysis is a condition that causes episodes of extreme muscle weakness typically beginning in childhood or adolescence. Most often, these episodes involve a temporary inability to move muscles in the arms and legs. Attacks cause severe weakness or paralysis that usually lasts from hours to days. Some people may have episodes almost every day, while others experience them weekly, monthly, or only rarely. Attacks can occur without warning or can be triggered by factors such as rest after exercise, a viral illness, or certain medications. Often, a large, carbohydrate-rich meal or vigorous exercise in the evening can trigger an attack upon waking the following morning. Although affected individuals usually regain their muscle strength between attacks, repeated episodes can lead to persistent muscle weakness later in life.
People with hypokalemic periodic paralysis have reduced levels of potassium in their blood (hypokalemia) during episodes of muscle weakness. Researchers are investigating how low potassium levels may be related to the muscle abnormalities in this condition.
How common is hypokalemic periodic paralysis?
Although its exact prevalence is unknown, hypokalemic periodic paralysis is estimated to affect 1 in 100,000 people. Men tend to experience symptoms of this condition more often than women.
Diagnosis/testing. The diagnosis of HOKPP is based on a history of episodes of flaccid paralysis; low serum concentration of potassium (<0.9 to 3.0 mmol/L) during attacks; the absence of myotonia clinically and on electromyography (EMG) (with the exception of one family with heat-induced myotonia and cold-induced HOKPP); and a family history consistent with autosomal dominant inheritance. Molecular genetic testing identifies disease-causing mutations in CACNA1S or SCN4A in 80% of individuals meeting clinical diagnostic criteria. Of all individuals with HOKPP, about 55-70% have mutations in CACNA1S and about 8-10% in SCN4A. Such testing is clinically available.
Management. Treatment of a paralytic crisis by administration of potassium by mouth or IV aims to normalize the serum concentration of potassium and to shorten the paralytic episode. ECG and blood potassium concentration must be monitored during treatment. Surveillance depends on the affected individual's symptoms and response to preventive treatment. Neurologic examination should focus on muscle strength in the legs to detect permanent weakness associated with myopathy. A number of factors can trigger paralytic attacks: unusually strenuous effort, excess of carbohydrate-rich meals, sweets, and alcohol should be avoided; oral or intravenous corticosteroids should be used with care; glucose infusion should be replaced by another type of infusion.
Potassium in doses of 0.2 to 0.4 mmol/kg is administered orally every 15 to 30 minutes over one to three hours.
If the individual is unable to swallow or does not tolerate potassium by mouth, potassium may be administered intravenously. In that case, it must be diluted in 5% mannitol rather than in glucose or sodium chloride, which trigger crises in individuals with HOKPP. The concentration of potassium administered intravenously must not exceed 40 mmol/L and the flow must not exceed 20 mmol/hour or 200-250 mmol/day; administration must be stopped when the serum potassium concentration is normalized, even if weakness persists.
Because the hypokalemia and subsequent changes in potassium
Genetic counseling. HOKPP is inherited in an autosomal dominant manner. Most individuals diagnosed with HOKPP have an affected parent. The proportion of cases caused by a de novo gene mutation is unknown. Offspring of a proband have a 50% risk of inheriting the mutation. Penetrance is about 90% in males and may be as low as 50% in females depending on the causative mutation. Prenatal testing is possible if the disease-causing mutation has been identified in the proband; however, requests for prenatal testing for conditions such as HOKPP that do not affect intellect and have some treatment available are not common.
POTASSIUM
Treatment of genetic diseases in tha real world 8: Hemochromatosis
Disease characteristics. HFE-associated hereditary hemochromatosis (HFE-HHC) is characterized by inappropriately high absorption of iron by the gastrointestinal mucosa, resulting in excessive storage of iron particularly in the liver, skin, pancreas, heart, joints, and testes. Abdominal pain, weakness, lethargy, and weight loss are early symptoms. Without therapy, males may develop symptoms between age 40 and 60 years and females after menopause. Hepatic fibrosis or cirrhosis may occur in untreated individuals after age 40 years. Other findings in untreated individuals may include progressive increase in skin pigmentation, diabetes mellitus, congestive heart failure and/or arrhythmias, arthritis, and hypogonadism.--> -->This description applies to individuals with clinical expression of HFE-HHC. A large, but yet as undefined, fraction of homozygotes for HFE-HHC do not develop clinical symptoms (i.e., penetrance is low).
Diagnosis/testing. The diagnosis of HFE-HHC in individuals with clinical symptoms consistent with HFE-HHC and/or biochemical evidence of iron overload is typically based on the results of the screening tests transferrin-iron saturation and serum ferritin concentration, and of confirmatory tests such as molecular genetic testing for the p.C282Y and p.H63D mutations in the HFE gene and/or histologic assessment of hepatic iron stores on liver biopsy. A threshold transferrin-iron saturation of 45% may be more sensitive for detecting HFE-HHC than the higher values used in the past. Although serum ferritin concentration may increase progressively over time in untreated individuals with HFE-HHC, it is not specific for HFE-HHC and cannot be used alone for identification of individuals with HFE-HHC. About 87% of individuals of European origin with HFE-HHC are either homozygotes for the p.C282Y mutation or compound heterozygotes for the p.C282Y and p.H63D mutations.
Management. Evaluations at initial diagnosis: liver biopsy in individuals with serum ferritin concentration greater than 1000 ng/mL to determine if cirrhosis is present. Treatment of manifestations: There is no general agreement that phlebotomy (removal of blood) treatment is indicated in the presence of biochemically defined abnormalities (i.e., elevated transferrin-iron saturation and elevated serum ferritin concentration) and the absence of characteristic clinical endpoints (i.e., diabetes mellitus, cirrhosis, and liver carcinoma). Since the long-term clinical course appears benign in the majority of those who have abnormal laboratory tests only, phlebotomy may be deferred; biannual follow-up testing for increasingly abnormal serum ferritin concentration and transferrin-iron saturation levels is recommended. In the presence of characteristic clinical endpoints, treatment by phlebotomy is indicated to maintain serum ferritin concentration at 50 ng/mL or lower. If affected individuals are identified before hepatic cirrhosis develops and if total body iron depletion is successfully accomplished by therapeutic phlebotomy, life expectancy approaches normal.
Genetic counseling. HFE-HHC is inherited in an autosomal recessive manner. Usually the genetic risk to sibs of a proband of having HFE-HHC is 25%. However, the high carrier frequency for a mutant HFE allele in the general population of European origin (11% of the population, or 1/9 persons) means that on occasion one parent has two abnormal HFE alleles, usually in the absence of clinical findings. In such instances, the risk to each sib of a proband of being homozygous for HFE-HHC is 50%. Offspring of an individual with HFE-HHC inherit one mutant HFE allele from the affected parent. Because the chance that the other parent is a carrier for a mutant HFE allele is 1/9, the risk to the offspring of having HFE-HHC is about 5%. Although prenatal testing would be technically feasible when both parents carry identified HFE mutations, such requests would be highly unusual because HFE-HHC is an adult-onset, treatable disease and the homozygous p.C282Y mutation has low clinical penetrance.
THERAPEUTIC PHLEBOTOMY
Diagnosis/testing. The diagnosis of HFE-HHC in individuals with clinical symptoms consistent with HFE-HHC and/or biochemical evidence of iron overload is typically based on the results of the screening tests transferrin-iron saturation and serum ferritin concentration, and of confirmatory tests such as molecular genetic testing for the p.C282Y and p.H63D mutations in the HFE gene and/or histologic assessment of hepatic iron stores on liver biopsy. A threshold transferrin-iron saturation of 45% may be more sensitive for detecting HFE-HHC than the higher values used in the past. Although serum ferritin concentration may increase progressively over time in untreated individuals with HFE-HHC, it is not specific for HFE-HHC and cannot be used alone for identification of individuals with HFE-HHC. About 87% of individuals of European origin with HFE-HHC are either homozygotes for the p.C282Y mutation or compound heterozygotes for the p.C282Y and p.H63D mutations.
Management. Evaluations at initial diagnosis: liver biopsy in individuals with serum ferritin concentration greater than 1000 ng/mL to determine if cirrhosis is present. Treatment of manifestations: There is no general agreement that phlebotomy (removal of blood) treatment is indicated in the presence of biochemically defined abnormalities (i.e., elevated transferrin-iron saturation and elevated serum ferritin concentration) and the absence of characteristic clinical endpoints (i.e., diabetes mellitus, cirrhosis, and liver carcinoma). Since the long-term clinical course appears benign in the majority of those who have abnormal laboratory tests only, phlebotomy may be deferred; biannual follow-up testing for increasingly abnormal serum ferritin concentration and transferrin-iron saturation levels is recommended. In the presence of characteristic clinical endpoints, treatment by phlebotomy is indicated to maintain serum ferritin concentration at 50 ng/mL or lower. If affected individuals are identified before hepatic cirrhosis develops and if total body iron depletion is successfully accomplished by therapeutic phlebotomy, life expectancy approaches normal.
Genetic counseling. HFE-HHC is inherited in an autosomal recessive manner. Usually the genetic risk to sibs of a proband of having HFE-HHC is 25%. However, the high carrier frequency for a mutant HFE allele in the general population of European origin (11% of the population, or 1/9 persons) means that on occasion one parent has two abnormal HFE alleles, usually in the absence of clinical findings. In such instances, the risk to each sib of a proband of being homozygous for HFE-HHC is 50%. Offspring of an individual with HFE-HHC inherit one mutant HFE allele from the affected parent. Because the chance that the other parent is a carrier for a mutant HFE allele is 1/9, the risk to the offspring of having HFE-HHC is about 5%. Although prenatal testing would be technically feasible when both parents carry identified HFE mutations, such requests would be highly unusual because HFE-HHC is an adult-onset, treatable disease and the homozygous p.C282Y mutation has low clinical penetrance.
THERAPEUTIC PHLEBOTOMY
Treament of genetic diseases in a real world 7: Wilson disease
See clinical synopsis in this link: wilson disease
Wilson disease can be treated effectively with metal chelating agent:
d-penicillamine (Cuprimine) especially in mild or asymptomatic cases, but patients with neurological symptoms way be worsen in the early period after treatment, and trientine(Syprine )is recommended for acute neurological alterations. Long term side effects of d-penicillamine is nephrotic syndrome, skin disruption and immune and bone marrow suppression. Zinc suphate is used in asymptomatic case or in maintenance phase after d-penicillamine treatment.
Agents/Circumstances to Avoid
Foods very high in copper (liver, brain, chocolate, mushrooms, shellfish, nuts), especially at the beginning of treatment
Wilson disease can be treated effectively with metal chelating agent:
d-penicillamine (Cuprimine) especially in mild or asymptomatic cases, but patients with neurological symptoms way be worsen in the early period after treatment, and trientine(Syprine )is recommended for acute neurological alterations. Long term side effects of d-penicillamine is nephrotic syndrome, skin disruption and immune and bone marrow suppression. Zinc suphate is used in asymptomatic case or in maintenance phase after d-penicillamine treatment.
Agents/Circumstances to Avoid
Foods very high in copper (liver, brain, chocolate, mushrooms, shellfish, nuts), especially at the beginning of treatment
Treatment of genetic diseases in a real world 6: Maturity Onset of Diabetes in the young (MODY)
WHAT IS MATURITY-ONSET DIABETES OF THE YOUNG (MODY)?
Maturity-Onset Diabetes of the Young or MODY affects 1-2% of people with diabetes, although it often goes unrecognised.
The 3 main features of MODY are:
Diabetes often develops before the age of 25
Diabetes runs in families from one generation to the next
Diabetes may be treated by diet or tablets and does not always need insulin treatment
WHY DOES MODY RUN IN FAMILIES?
MODY runs in families because of a change in a single gene which is passed on by affected parents to their children. We call this Autosomal Dominant Inheritance. All children of an affected parent with MODY have a 50% chance of inheriting the affected gene and developing MODY themselves.
WHY IS IT IMPORTANT TO RECOGNISE IT?
There are different types of MODY. By finding out which type of MODY a person has the most appropriate treatment for them can be determined.
Knowing the type of MODY a person has also means we can advise them about how their diabetes will progress in the future.
As it runs in families, it is important to advise other family members of their risk of inheriting it.
WHAT DIFFERENT TYPES OF MODY HAVE BEEN IDENTIFIED?
MODY is caused by a change in a single gene. 6 genes have been identified that account for 87% of UK MODY:
HNF1-a
Treatment for patients with HNF1-a
Patients with HNF-1a MODY are extremely sensitive to the blood sugar lowering effects of a group of drugs called sulphonylureas (SU). This is an example of pharmacogenetics in diabetes – a persons genes influencing response to treatment. SUs include drugs like Gliclazide, Glipizide, Glibenclamide, Tolbutamide. SUs work to stimulate the pancreas to produce insulin. Preliminary findings are that SU sensitivity in HNF-1a MODY is due to two factors: Firstly, an increased pancreatic response to SUs, and secondly an increased sensitivity to insulin compared with Type 2 diabetes.
Glucokinase
HNF1-b (including Renal Cysts and Diabetes (RCAD)
HNF4-a
IPF1
Neuro D1
Changes in these different genes lead to different types of MODY. There are still more genes to identify as 13% of MODY is not yet accounted for.
http://www.projects.ex.ac.uk/diabetesgenes/mody/ATHtalk.PPT
Maturity-Onset Diabetes of the Young or MODY affects 1-2% of people with diabetes, although it often goes unrecognised.
The 3 main features of MODY are:
Diabetes often develops before the age of 25
Diabetes runs in families from one generation to the next
Diabetes may be treated by diet or tablets and does not always need insulin treatment
WHY DOES MODY RUN IN FAMILIES?
MODY runs in families because of a change in a single gene which is passed on by affected parents to their children. We call this Autosomal Dominant Inheritance. All children of an affected parent with MODY have a 50% chance of inheriting the affected gene and developing MODY themselves.
WHY IS IT IMPORTANT TO RECOGNISE IT?
There are different types of MODY. By finding out which type of MODY a person has the most appropriate treatment for them can be determined.
Knowing the type of MODY a person has also means we can advise them about how their diabetes will progress in the future.
As it runs in families, it is important to advise other family members of their risk of inheriting it.
WHAT DIFFERENT TYPES OF MODY HAVE BEEN IDENTIFIED?
MODY is caused by a change in a single gene. 6 genes have been identified that account for 87% of UK MODY:
HNF1-a
Treatment for patients with HNF1-a
Patients with HNF-1a MODY are extremely sensitive to the blood sugar lowering effects of a group of drugs called sulphonylureas (SU). This is an example of pharmacogenetics in diabetes – a persons genes influencing response to treatment. SUs include drugs like Gliclazide, Glipizide, Glibenclamide, Tolbutamide. SUs work to stimulate the pancreas to produce insulin. Preliminary findings are that SU sensitivity in HNF-1a MODY is due to two factors: Firstly, an increased pancreatic response to SUs, and secondly an increased sensitivity to insulin compared with Type 2 diabetes.
Glucokinase
HNF1-b (including Renal Cysts and Diabetes (RCAD)
HNF4-a
IPF1
Neuro D1
Changes in these different genes lead to different types of MODY. There are still more genes to identify as 13% of MODY is not yet accounted for.
http://www.projects.ex.ac.uk/diabetesgenes/mody/ATHtalk.PPT
Wednesday, 21 November 2007
Treatment of genetic diseases: in the real world. 5. Marfan syndrome
Marfan Syndrome is a multisystem inherited diseases; the major organ involvement is cardiovascular system. patient s with Marfan Syndrome will gradually develop enlargment of the great artery of heart (Aorta) due to weakness of the supporting tissues and leading to tearing (dissection) which are mostly fatal. See more informations of Marfan Syndrome in the link provided
http://clinicalgenetics.blogspot.com/2007/11/marfan-syndrome.html
Treatment of Marfan Syndrome is including advice to avoid vigorous exercise and regularly taking drug which reduce the force of heart beat that are proved to slow progression of the disease. Echocardiography (Ultrasound image of the heart and arteries) periodically to measure sizes of arteries in case of too much enlarge that hhaving high risk for tearing or rupture, prophylactic surgery will have done.
Drug use in Marfan Syndrome
Propanolol Inderal from Wyeth NEngl J Med. 1994 May 12;330(19):1335-41
Enalapril Renitec from Merck Am J Cardiol 2005;95:1125–1127
Atenolo and losartan
A Clinical Trial Comparing Atenolol and Losartan Is Beginning
Reed E. Pyeritz, M.D., Ph.D.
Professor of Medicine & Genetics
University of Pennsylvania School of Medicine
Philadelphia, PA, USA
http://clinicalgenetics.blogspot.com/2007/11/marfan-syndrome.html
Treatment of Marfan Syndrome is including advice to avoid vigorous exercise and regularly taking drug which reduce the force of heart beat that are proved to slow progression of the disease. Echocardiography (Ultrasound image of the heart and arteries) periodically to measure sizes of arteries in case of too much enlarge that hhaving high risk for tearing or rupture, prophylactic surgery will have done.
Drug use in Marfan Syndrome
Propanolol Inderal from Wyeth NEngl J Med. 1994 May 12;330(19):1335-41
Enalapril Renitec from Merck Am J Cardiol 2005;95:1125–1127
Atenolo and losartan
A Clinical Trial Comparing Atenolol and Losartan Is Beginning
Reed E. Pyeritz, M.D., Ph.D.
Professor of Medicine & Genetics
University of Pennsylvania School of Medicine
Philadelphia, PA, USA
Treatment of genetic diseases: in the real world.4. klinefelter's syndrome
What is Klinefelter syndrome?
Klinefelter syndrome is a chromosomal condition that affects male sexual development. Most males with Klinefelter syndrome have one extra copy of the X chromosome in each cell. Because their testicles do not develop normally, affected males may have low levels of the hormone testosterone beginning during puberty. A lack of this hormone can lead to breast development (gynecomastia), reduced facial and body hair, and an inability to father children (infertility). Compared with other men, adult males with Klinefelter syndrome have an increased risk of developing breast cancer and a chronic inflammatory disease called systemic lupus erythematosus. Their chance of developing these disorders is similar to that of normal adult females.
Boys with Klinefelter syndrome may have learning disabilities and difficulty with speech and language development. They tend to be quiet, sensitive, and unassertive, but personality characteristics vary among males with this condition.
Variants of Klinefelter syndrome, which involve more than one extra X chromosome or extra copies of both the X and Y chromosomes in each cell, tend to have more severe signs and symptoms. These disorders affect male sexual development and are associated with decreased IQ, distinctive facial features, skeletal abnormalities, poor coordination, and severe problems with speech.
How common is Klinefelter syndrome?
Klinefelter syndrome affects 1 in 500 to 1,000 males. Variants of Klinefelter syndrome are much rarer, occurring in 1 in 50,000 or fewer male births. Females are not affected by Klinefelter syndrome
Initial treatment
Treatment for Klinefelter syndrome usually starts when a boy is about 11 to 12 years old. It begins with measuring the amount of testosterone and other hormones in his blood. Many teenage boys with Klinefelter syndrome don't have low levels of testosterone.4 If a boy's testosterone level is low, he is given a man-made form of testosterone (Depo-Testosterone) on a regular basis. Testosterone can be given as an injection or through a skin patch or gel.
Testosterone:
Increases body hair, mainly on the face (beard), under the arm (axillary), and in the genital area (pubic).
Increases muscle development.
Increases sex drive.
Helps prevent osteoporosis.
May prevent or shrink enlarged breasts.
Provides better self-esteem by allowing the boy to "fit in" with his peers. This can result in more successful interpersonal relationships.
Side effects of testosterone replacement therapy are uncommon, but include worsening acne, overly rapid sexual development, and behavior problems (such as being overly aggressive). Careful monitoring is important to prevent these side effects.
Ongoing treatment
Ongoing treatment forKlinefelter syndrome may include:
Help for language and learning difficulties. If language delays are identified in early childhood, educational assistance and speech therapy can be used to treat the problem. Children with learning difficulties can receive educational support through the school system.
Testosterone replacement. This is given by injection or through a skin patch or gel. Testosterone replacement usually continues throughout the man's life but does not help infertility. For men who want to start a family, counseling and treatment at a fertility clinic are generally recommended.
Regular medical checkups to monitor for development of other conditions, such as autoimmune diseases, behavior problems, or psychiatric disorders, such as depression. Professional counseling or medication may be needed.
DEPO-TESTOSTERONE from Pfizer
Klinefelter syndrome is a chromosomal condition that affects male sexual development. Most males with Klinefelter syndrome have one extra copy of the X chromosome in each cell. Because their testicles do not develop normally, affected males may have low levels of the hormone testosterone beginning during puberty. A lack of this hormone can lead to breast development (gynecomastia), reduced facial and body hair, and an inability to father children (infertility). Compared with other men, adult males with Klinefelter syndrome have an increased risk of developing breast cancer and a chronic inflammatory disease called systemic lupus erythematosus. Their chance of developing these disorders is similar to that of normal adult females.
Boys with Klinefelter syndrome may have learning disabilities and difficulty with speech and language development. They tend to be quiet, sensitive, and unassertive, but personality characteristics vary among males with this condition.
Variants of Klinefelter syndrome, which involve more than one extra X chromosome or extra copies of both the X and Y chromosomes in each cell, tend to have more severe signs and symptoms. These disorders affect male sexual development and are associated with decreased IQ, distinctive facial features, skeletal abnormalities, poor coordination, and severe problems with speech.
How common is Klinefelter syndrome?
Klinefelter syndrome affects 1 in 500 to 1,000 males. Variants of Klinefelter syndrome are much rarer, occurring in 1 in 50,000 or fewer male births. Females are not affected by Klinefelter syndrome
Initial treatment
Treatment for Klinefelter syndrome usually starts when a boy is about 11 to 12 years old. It begins with measuring the amount of testosterone and other hormones in his blood. Many teenage boys with Klinefelter syndrome don't have low levels of testosterone.4 If a boy's testosterone level is low, he is given a man-made form of testosterone (Depo-Testosterone) on a regular basis. Testosterone can be given as an injection or through a skin patch or gel.
Testosterone:
Increases body hair, mainly on the face (beard), under the arm (axillary), and in the genital area (pubic).
Increases muscle development.
Increases sex drive.
Helps prevent osteoporosis.
May prevent or shrink enlarged breasts.
Provides better self-esteem by allowing the boy to "fit in" with his peers. This can result in more successful interpersonal relationships.
Side effects of testosterone replacement therapy are uncommon, but include worsening acne, overly rapid sexual development, and behavior problems (such as being overly aggressive). Careful monitoring is important to prevent these side effects.
Ongoing treatment
Ongoing treatment forKlinefelter syndrome may include:
Help for language and learning difficulties. If language delays are identified in early childhood, educational assistance and speech therapy can be used to treat the problem. Children with learning difficulties can receive educational support through the school system.
Testosterone replacement. This is given by injection or through a skin patch or gel. Testosterone replacement usually continues throughout the man's life but does not help infertility. For men who want to start a family, counseling and treatment at a fertility clinic are generally recommended.
Regular medical checkups to monitor for development of other conditions, such as autoimmune diseases, behavior problems, or psychiatric disorders, such as depression. Professional counseling or medication may be needed.
DEPO-TESTOSTERONE from Pfizer
Treatment of genetic diseases: in the real world. 3. Turner syndrome
What is Turner syndrome?
Turner syndrome is a chromosomal condition that alters development in females. Women with this condition tend to be shorter than average and are usually unable to conceive a child (infertile) because of an absence of ovarian function. Other features of this condition that can vary among women who have Turner syndrome include: extra skin on the neck (webbed neck), puffiness or swelling (lymphedema) of the hands and feet, skeletal abnormalities, heart defects and kidney problems.
This condition occurs in about 1 in 2,500 female births worldwide, but is much more common among pregnancies that do not survive to term (miscarriages and stillbirths).
Turner syndrome is a chromosomal condition related to the X chromosome. Researchers have not yet determined which genes on the X chromosome are responsible for most signs and symptoms of Turner syndrome. They have, however, identified one gene called SHOX that is important for bone development and growth. Missing one copy of this gene likely causes short stature and skeletal abnormalities in women with Turner syndrome.
What is the treatment for Turner syndrome?
During childhood and adolescence, girls may be under the care of a pediatric endocrinologist, who is a specialist in childhood conditions of the hormones and metabolism.
Growth hormone injections are beneficial in some individuals with Turner syndrome. Injections often begin in early childhood and may increase final adult height by a few inches.
Estrogen replacement therapy is usually started at the time of normal puberty, around 12 years to start breast development. Estrogen and progesterone are given a little later to begin a monthly 'period,' which is necessary to keep the womb healthy. Estrogen is also given to prevent osteoporosis.
Babies born with a heart murmur or narrowing of the aorta may need surgery to correct the problem. A heart expert (cardiologist) will assess and follow up any treatment necessary.
Girls who have Turner syndrome are more likely to get middle ear infections. Repeated infections may lead to hearing loss and should be evaluated by the pediatrician. An ear, nose and throat specialist (ENT) may be involved in caring for this health issue.
High blood pressure is quite common in women who have Turner syndrome. In some cases, the elevated blood pressure is due to narrowing of the aorta or a kidney abnormality. However, most of the time, no specific cause for the elevation is identified. Blood pressure should be checked routinely and, if necessary, treated with medication. Women who have Turner syndrome have a slightly higher risk of having an under active thyroid or developing diabetes. This should also be monitored during routine health maintenance visits and treated if necessary.
Regular health checks are very important. Special clinics for the care of girls and women who have Turner syndrome are available in some areas, with access to a variety of specialists. Early preventive care and treatment is very important.
Almost all women are infertile, but pregnancy with donor embryos may be possible.
Drug used for Turner syndrome patients
Estrogen to increase height, produce secondary sexual charactristics, prevent osteoporois, improve fertility in some cases.
Premarin conjugated estrogen from Wyeth
Growth hormone to increase height.
Humatrope recombinant Human Growth Hormone from EliLily
Thyroid hormone in patients with hypothyroids
Eltroxin T4 Tetrathyroiodine from Glaxo Welcome
Turner syndrome is a chromosomal condition that alters development in females. Women with this condition tend to be shorter than average and are usually unable to conceive a child (infertile) because of an absence of ovarian function. Other features of this condition that can vary among women who have Turner syndrome include: extra skin on the neck (webbed neck), puffiness or swelling (lymphedema) of the hands and feet, skeletal abnormalities, heart defects and kidney problems.
This condition occurs in about 1 in 2,500 female births worldwide, but is much more common among pregnancies that do not survive to term (miscarriages and stillbirths).
Turner syndrome is a chromosomal condition related to the X chromosome. Researchers have not yet determined which genes on the X chromosome are responsible for most signs and symptoms of Turner syndrome. They have, however, identified one gene called SHOX that is important for bone development and growth. Missing one copy of this gene likely causes short stature and skeletal abnormalities in women with Turner syndrome.
What is the treatment for Turner syndrome?
During childhood and adolescence, girls may be under the care of a pediatric endocrinologist, who is a specialist in childhood conditions of the hormones and metabolism.
Growth hormone injections are beneficial in some individuals with Turner syndrome. Injections often begin in early childhood and may increase final adult height by a few inches.
Estrogen replacement therapy is usually started at the time of normal puberty, around 12 years to start breast development. Estrogen and progesterone are given a little later to begin a monthly 'period,' which is necessary to keep the womb healthy. Estrogen is also given to prevent osteoporosis.
Babies born with a heart murmur or narrowing of the aorta may need surgery to correct the problem. A heart expert (cardiologist) will assess and follow up any treatment necessary.
Girls who have Turner syndrome are more likely to get middle ear infections. Repeated infections may lead to hearing loss and should be evaluated by the pediatrician. An ear, nose and throat specialist (ENT) may be involved in caring for this health issue.
High blood pressure is quite common in women who have Turner syndrome. In some cases, the elevated blood pressure is due to narrowing of the aorta or a kidney abnormality. However, most of the time, no specific cause for the elevation is identified. Blood pressure should be checked routinely and, if necessary, treated with medication. Women who have Turner syndrome have a slightly higher risk of having an under active thyroid or developing diabetes. This should also be monitored during routine health maintenance visits and treated if necessary.
Regular health checks are very important. Special clinics for the care of girls and women who have Turner syndrome are available in some areas, with access to a variety of specialists. Early preventive care and treatment is very important.
Almost all women are infertile, but pregnancy with donor embryos may be possible.
Drug used for Turner syndrome patients
Estrogen to increase height, produce secondary sexual charactristics, prevent osteoporois, improve fertility in some cases.
Premarin conjugated estrogen from Wyeth
Growth hormone to increase height.
Humatrope recombinant Human Growth Hormone from EliLily
Thyroid hormone in patients with hypothyroids
Eltroxin T4 Tetrathyroiodine from Glaxo Welcome
Treatment of genetic diseases: in the real world. 2. Hemophilia
Hemophilia is an inherited bleeding disorders that was record since the ancient time.
The male new born has to be circumscribed for religious purpose, the affected individuals can be bleed until dead leading to exception for families with history of bleeding in male.
The most famous family with hemophilia is the Queen victorian royal family; there are many male suffered from this kind of severe bleeding disorders.
Once upon a time, those affected individulas died in before 10 years olf from sever intracranila bleeding. Nowadays, we know that these patients are lacking of clotting factor in their blood. Two common types of disease lack from differnet factor: factor 8 in hemophilia A and factor 9 in hemophilia B. These factors are found in normal individual plasma and that can be collected, concentrate and given to patiens in need. from the advance of genetic engineering, we can synthesized factor 8 or 9 with recombinant technology to produce large amount of factors independently for blood donors anymore, but they are still expensive and restricted for use in the most severe one.
These are the list of saving life products for hemophiliac patients:
Fresh Frozen Plasma
Cryoprecipitated (factor 8 rich)
Cryoremoved plasma (factor 9 rich)
Factor 7 recombinant: Novoseven from Novo Nordisk (can be used in both types)
Factor 8 recombinant: Advate from baxter, Helixate from Behring
Factor 9 recombinant: BeneFIX from Wyeth
Anti-Inhibitor Coagulant Complex FEIBA from Baxter (can be used in both types)
In mild hemophilia A or hemophiliac patients undegoing to perform oral procedure: transamin (tranexamic acid) from Daiichi
The male new born has to be circumscribed for religious purpose, the affected individuals can be bleed until dead leading to exception for families with history of bleeding in male.
The most famous family with hemophilia is the Queen victorian royal family; there are many male suffered from this kind of severe bleeding disorders.
Once upon a time, those affected individulas died in before 10 years olf from sever intracranila bleeding. Nowadays, we know that these patients are lacking of clotting factor in their blood. Two common types of disease lack from differnet factor: factor 8 in hemophilia A and factor 9 in hemophilia B. These factors are found in normal individual plasma and that can be collected, concentrate and given to patiens in need. from the advance of genetic engineering, we can synthesized factor 8 or 9 with recombinant technology to produce large amount of factors independently for blood donors anymore, but they are still expensive and restricted for use in the most severe one.
These are the list of saving life products for hemophiliac patients:
Fresh Frozen Plasma
Cryoprecipitated (factor 8 rich)
Cryoremoved plasma (factor 9 rich)
Factor 8 concentrate plasma
Factor 9 concentrate plasma
Factor 7 recombinant: Novoseven from Novo Nordisk (can be used in both types)
Factor 8 recombinant: Advate from baxter, Helixate from Behring
Factor 9 recombinant: BeneFIX from Wyeth
Anti-Inhibitor Coagulant Complex FEIBA from Baxter (can be used in both types)
In mild hemophilia A or hemophiliac patients undegoing to perform oral procedure: transamin (tranexamic acid) from Daiichi
Treatment of genetic diseases: in the real world. 1. familial hypercholesterolemia
One of the most common misunderstanding or negative attitudes towards genetic diseases is Genetic diseases can not be treated. They may sometimes hear about gene therapy, cloning or other advanced knowledge requirement to understand, so they leave genetics at the corner of their mind. But what do genetics doctors say?
I have to say that there are thousands of genetic diseases (and this long list is growing rapidly) that are treated or can be prevented with drugs, diet therapy, or surgical procedures effectively.
For emphasizing this promising areas of medicine, I will list these genetic diseases and their management that change their hopeless life to the bright new one.
The first one I'd like to mention is familial hypercholesterolemia.
Familial hypercholesterolemia
Familial hypercholesterolemia is one of the most common genetic diseases in the world; the prevalence of heterozygous (get one abnormal gene from only one parent) affected people is about 1 in 500, which equal to prevalence of stroke in England.
Affected people have consistenly high levels of low density lipoprotein (LDL or bad cholesterol) which leads to premature atherosclerosis of the coronary arteries, cerebral arteries and peripheral vessels. Typically in affected men, heart attacks occur in their 40s to 50s, and 85% of men with this disorder have experienced a heart attack by age 60. The incidence of heart attacks in women with this disorder is also increased, but happens 10 years later than in men.
In homozygous (receive abnormal genes from both parents) individuals the condition is more severe, and cholesterol values may exceed 600 mg/dl (normal not exceed 200 mg/dl) These individuals develop waxy plaques (xanthoma) beneath the skin over their elbows, knees, buttocks. These are deposits of cholesterol in the skin. In addition, they develop deposits in tendons, and around the cornea of the eye. atherosclerosis begins before pubery and heart attacks and death may occur before age30, or they may require higly invasive surgery such as a liver transplant. Fortunately homozygous individulas are extremely rare in about 1 in million.
Hetrozygous patients may respond well to diet modifications combined with strong lipid lowering agents which have a class name statins.
Statins are the most common and one of the most prescribed drugs nowadays for treatment of high blood cholesterol. Most of high blood cholesterol individuals are partially inherited, but not in the case of familial hypercholesterolmeia which transfer to the next generation at rate about 50 %.
There are many guidelines for managing affected individulas.
Statins provides excellent control of inherited high cholestrol levels. The statins work by reducing the manufacture of cholestrol by cells. This stimulated LDL receptor gene expression. As a result, the receptors produced by the normal gene wil reduce cholesterol levels. the effect of statins can be enhanced by bile acid sequestants or cholesterol absorption inhibitors such as plant sterols or the recently introduced drug, ezetimibe.
Many patients with FH can achieve target cholesterol levels.
STATIN TREATMENT OF FH MALES IS ONE OF HE MOST COST_EFFECTIVE MEDICAL INTERVENTIONS AVAILABLE AND SEVERAL LINE OF EVIDENCE POINT TOWARDS MAJOR IMPROVEMENTS IN CARDIOVASCULAR EVENT RATES AND TOTAL MORTALITY(DEAD) OF FH PATIENTS.
These are the names of drugs can be used for Familail Hypercholesterolemia:
Lipitor from Pfizer : Atorvastatin
Crestor from Aventis: Rosuvastatin
high dose Zocor from Merck: simvastatin
Ezetrol from Merck: ezetimibe in combination with Zocor
I have to say that there are thousands of genetic diseases (and this long list is growing rapidly) that are treated or can be prevented with drugs, diet therapy, or surgical procedures effectively.
For emphasizing this promising areas of medicine, I will list these genetic diseases and their management that change their hopeless life to the bright new one.
The first one I'd like to mention is familial hypercholesterolemia.
Familial hypercholesterolemia
Familial hypercholesterolemia is one of the most common genetic diseases in the world; the prevalence of heterozygous (get one abnormal gene from only one parent) affected people is about 1 in 500, which equal to prevalence of stroke in England.
Affected people have consistenly high levels of low density lipoprotein (LDL or bad cholesterol) which leads to premature atherosclerosis of the coronary arteries, cerebral arteries and peripheral vessels. Typically in affected men, heart attacks occur in their 40s to 50s, and 85% of men with this disorder have experienced a heart attack by age 60. The incidence of heart attacks in women with this disorder is also increased, but happens 10 years later than in men.
In homozygous (receive abnormal genes from both parents) individuals the condition is more severe, and cholesterol values may exceed 600 mg/dl (normal not exceed 200 mg/dl) These individuals develop waxy plaques (xanthoma) beneath the skin over their elbows, knees, buttocks. These are deposits of cholesterol in the skin. In addition, they develop deposits in tendons, and around the cornea of the eye. atherosclerosis begins before pubery and heart attacks and death may occur before age30, or they may require higly invasive surgery such as a liver transplant. Fortunately homozygous individulas are extremely rare in about 1 in million.
Hetrozygous patients may respond well to diet modifications combined with strong lipid lowering agents which have a class name statins.
Statins are the most common and one of the most prescribed drugs nowadays for treatment of high blood cholesterol. Most of high blood cholesterol individuals are partially inherited, but not in the case of familial hypercholesterolmeia which transfer to the next generation at rate about 50 %.
There are many guidelines for managing affected individulas.
Statins provides excellent control of inherited high cholestrol levels. The statins work by reducing the manufacture of cholestrol by cells. This stimulated LDL receptor gene expression. As a result, the receptors produced by the normal gene wil reduce cholesterol levels. the effect of statins can be enhanced by bile acid sequestants or cholesterol absorption inhibitors such as plant sterols or the recently introduced drug, ezetimibe.
Many patients with FH can achieve target cholesterol levels.
STATIN TREATMENT OF FH MALES IS ONE OF HE MOST COST_EFFECTIVE MEDICAL INTERVENTIONS AVAILABLE AND SEVERAL LINE OF EVIDENCE POINT TOWARDS MAJOR IMPROVEMENTS IN CARDIOVASCULAR EVENT RATES AND TOTAL MORTALITY(DEAD) OF FH PATIENTS.
These are the names of drugs can be used for Familail Hypercholesterolemia:
Lipitor from Pfizer : Atorvastatin
Crestor from Aventis: Rosuvastatin
high dose Zocor from Merck: simvastatin
Ezetrol from Merck: ezetimibe in combination with Zocor
Genetics and hearing impairment
Hearing impairment is the decreased ability to hear and discriminate among sounds. It is one of the most common birth defects. Each year in the United States, about 12,000 babies (3 in 1,000) are born with significant hearing impairment (1). Hearing impairment that is present at birth is called congenital hearing impairment. Hearing impairment also can develop later in childhood or during adulthood.
The Centers for Disease Control and Prevention (CDC) recommends that all babies be screened for hearing impairment before 1 month of age, preferably before they leave the hospital (1). This is because language and communication develop rapidly during the first two to three years of life, and undetected hearing impairment can lead to delays in developing these skills. Without newborn screening, children with hearing impairment usually are not diagnosed until 2 to 3 years of age.
The goal of early screening, diagnosis and treatment is to help children with hearing impairment develop language and academic skills equal to those of their peers. Most states have an Early Hearing Detection and Intervention Program to help ensure that all babies are screened, and that infants who do not pass the screening receive the follow-up care they need. The March of Dimes, the American Academy of Pediatrics, the Maternal and Child Health Bureau, the CDC and others strongly support these programs.
What causes hearing impairment in babies and children?Hearing impairment can be inherited (genetic) or nongenetic. Nongenetic causes include illness or injury occurring before, during or after birth. In some cases, the cause of hearing impairment is not known. About 90 percent of babies with congenital hearing impairment are born to hearing parents.
Genetic factors are believed to cause 33 percent of cases of hearing impairment in infants and young children. Scientists believe that mutations (changes) in as many as 400 genes may contribute to hearing impairment.
Genetic causes of hearing impairment can be:
Syndromatic: One feature of a group of birth defects that occur together. This type of impairment accounts for about 30 percent of cases.
Nonsyndromatic: A solitary birth defect. About 30 percent of cases of nonsyndromatic hearing impairment are caused by a mutation in a gene called Connexin 26.About one-third of cases of hearing impairment are caused by nongenetic factors. They include illnesses during pregnancy, such as:
Rubella (German measles)
Cytomegalovirus infections
Toxoplasmosis
Herpes infection
Syphilis
Preterm birth (before 37 completed weeks of pregnancy) also can be a cause.
After birth, head injuries and childhood infections (such as meningitis, measles or chickenpox) can cause permanent hearing impairment. Certain medications, such as the antibiotic streptomycin and related drugs, also can cause hearing impairment. Ear infection (otitis media) may cause temporary hearing impairment. Frequent and poorly treated ear infections can cause damage sufficient to impair hearing.
The causes of the remaining third of cases of hearing impairment in infants and children are unknown.
The Centers for Disease Control and Prevention (CDC) recommends that all babies be screened for hearing impairment before 1 month of age, preferably before they leave the hospital (1). This is because language and communication develop rapidly during the first two to three years of life, and undetected hearing impairment can lead to delays in developing these skills. Without newborn screening, children with hearing impairment usually are not diagnosed until 2 to 3 years of age.
The goal of early screening, diagnosis and treatment is to help children with hearing impairment develop language and academic skills equal to those of their peers. Most states have an Early Hearing Detection and Intervention Program to help ensure that all babies are screened, and that infants who do not pass the screening receive the follow-up care they need. The March of Dimes, the American Academy of Pediatrics, the Maternal and Child Health Bureau, the CDC and others strongly support these programs.
What causes hearing impairment in babies and children?Hearing impairment can be inherited (genetic) or nongenetic. Nongenetic causes include illness or injury occurring before, during or after birth. In some cases, the cause of hearing impairment is not known. About 90 percent of babies with congenital hearing impairment are born to hearing parents.
Genetic factors are believed to cause 33 percent of cases of hearing impairment in infants and young children. Scientists believe that mutations (changes) in as many as 400 genes may contribute to hearing impairment.
Genetic causes of hearing impairment can be:
Syndromatic: One feature of a group of birth defects that occur together. This type of impairment accounts for about 30 percent of cases.
Nonsyndromatic: A solitary birth defect. About 30 percent of cases of nonsyndromatic hearing impairment are caused by a mutation in a gene called Connexin 26.About one-third of cases of hearing impairment are caused by nongenetic factors. They include illnesses during pregnancy, such as:
Rubella (German measles)
Cytomegalovirus infections
Toxoplasmosis
Herpes infection
Syphilis
Preterm birth (before 37 completed weeks of pregnancy) also can be a cause.
After birth, head injuries and childhood infections (such as meningitis, measles or chickenpox) can cause permanent hearing impairment. Certain medications, such as the antibiotic streptomycin and related drugs, also can cause hearing impairment. Ear infection (otitis media) may cause temporary hearing impairment. Frequent and poorly treated ear infections can cause damage sufficient to impair hearing.
The causes of the remaining third of cases of hearing impairment in infants and children are unknown.
Genetics and miscarriage
Miscarriage is the loss of a pregnancy before 20 weeks gestation. There are many reasons for pregnancy loss. It is important to realize that in many cases, no cause for past miscarriage(s) is identified. Most causes of miscarriage are not under our control. It is important to remember that women who have a miscarriage(s) still have a good chance for a successful future pregnancy.
Genetic Causes
Fetal Chromosomal Abnormalities : Approximately 50% of first trimester miscarriages are due to a chromosome abnormality in the fetus. Chromosomes are the inherited structures in the cells of our bodies. There are 46 chromosomes in each cell, arranged into 23 pairs. A baby has two copies of every chromosome – one inherited from the mother in the egg, and the other inherited from the father in the sperm. Each chromosome holds hundreds to thousands of genes, which are responsible for growth and development. An extra chromosome or a missing chromosome can cause miscarriage, usually in the first or second trimester of pregnancy, or can lead to a child with learning difficulties or mental retardation and birth defects.Chromosome abnormalities involving a missing or extra chromosome are not inherited or caused by an exposure during pregnancy. Instead, they result from a chance mistake in cell division at the time of conception. This error is a random event that can occur in anyone’s pregnancy. Once a couple has had a pregnancy affected by a chromosome abnormality, there is a slightly greater chance for their future pregnancies to be affected with chromosome abnormalities. In some cases prenatal diagnosis, such as chorionic villus sampling (CVS) or amniocentesis , are offered in future pregnancies.
Inherited Chromosomal Rearrangements : An inherited problem with the chromosomes can also cause miscarriage. A parent can have a rearrangement (a "translocation") of his or her chromosomes, in which the chromosomes are structured differently. The parent should have no health problems because, although his or her chromosomes are rearranged, they are balanced – that is, there are no missing or extra pieces of the chromosomes. However, because of the way the chromosomes are passed from parent to child, the baby may inherit extra or missing pieces of a chromosome. Extra and missing genetic material lead to "chromosomal imbalance" and can cause mental retardation and birth defects in a liveborn or cause a miscarriage. For couples who have had multiple miscarriages, the chance that one of the parents has a chromosomal rearrangement is approximately 2-4%. While parents who carry chromosomal rearrangements are at increased risk to have further miscarriages or babies born with health problems, they can also produce healthy children. Chromosome studies can be performed on parents’ blood to see if either parent is a carrier of a chromosomal rearrangement.
Gene Mutation: Another genetic cause of miscarriage is a change (mutation) in a single gene (or several genes) on the chromosomes. This can cause specific genetic diseases or birth defects. Mutations can occur spontaneously in pregnancies or can be inherited from parents who themselves are healthy. Birth defects associated with these conditions can sometimes be detected during pregnancy by a sonogram. If there is a history of a specific disorder in a parent or family member, single gene disorders can be tested for prenatally in some cases.
Maternal Health Issues: Other reasons for pregnancy loss are related to maternal health. An abnormally shaped uterus can lead to pregnancy loss. Health problems such as hormonal imbalance, poorly-controlled diabetes, lupus and other immune system abnormalities, kidney and heart disease, and hypertension can create difficulties in carrying a pregnancy to term. These causes of miscarriage can be evaluated by blood tests and an ultrasound examination (sonogram) of the uterus. Your doctor can evaluate you for these problems. Environment: Another cause of pregnancy loss is an environmental exposure during pregnancy. For example, exposure to drugs, alcohol, or high levels of radiation can lead to miscarriage. Infections can cause miscarriage. The risk of miscarriage may be greater in women who smoke.
Remember, even when repeated miscarriages occur, there is a good chance of successful next pregnancy.
Genetic Causes
Fetal Chromosomal Abnormalities : Approximately 50% of first trimester miscarriages are due to a chromosome abnormality in the fetus. Chromosomes are the inherited structures in the cells of our bodies. There are 46 chromosomes in each cell, arranged into 23 pairs. A baby has two copies of every chromosome – one inherited from the mother in the egg, and the other inherited from the father in the sperm. Each chromosome holds hundreds to thousands of genes, which are responsible for growth and development. An extra chromosome or a missing chromosome can cause miscarriage, usually in the first or second trimester of pregnancy, or can lead to a child with learning difficulties or mental retardation and birth defects.Chromosome abnormalities involving a missing or extra chromosome are not inherited or caused by an exposure during pregnancy. Instead, they result from a chance mistake in cell division at the time of conception. This error is a random event that can occur in anyone’s pregnancy. Once a couple has had a pregnancy affected by a chromosome abnormality, there is a slightly greater chance for their future pregnancies to be affected with chromosome abnormalities. In some cases prenatal diagnosis, such as chorionic villus sampling (CVS) or amniocentesis , are offered in future pregnancies.
Inherited Chromosomal Rearrangements : An inherited problem with the chromosomes can also cause miscarriage. A parent can have a rearrangement (a "translocation") of his or her chromosomes, in which the chromosomes are structured differently. The parent should have no health problems because, although his or her chromosomes are rearranged, they are balanced – that is, there are no missing or extra pieces of the chromosomes. However, because of the way the chromosomes are passed from parent to child, the baby may inherit extra or missing pieces of a chromosome. Extra and missing genetic material lead to "chromosomal imbalance" and can cause mental retardation and birth defects in a liveborn or cause a miscarriage. For couples who have had multiple miscarriages, the chance that one of the parents has a chromosomal rearrangement is approximately 2-4%. While parents who carry chromosomal rearrangements are at increased risk to have further miscarriages or babies born with health problems, they can also produce healthy children. Chromosome studies can be performed on parents’ blood to see if either parent is a carrier of a chromosomal rearrangement.
Gene Mutation: Another genetic cause of miscarriage is a change (mutation) in a single gene (or several genes) on the chromosomes. This can cause specific genetic diseases or birth defects. Mutations can occur spontaneously in pregnancies or can be inherited from parents who themselves are healthy. Birth defects associated with these conditions can sometimes be detected during pregnancy by a sonogram. If there is a history of a specific disorder in a parent or family member, single gene disorders can be tested for prenatally in some cases.
Maternal Health Issues: Other reasons for pregnancy loss are related to maternal health. An abnormally shaped uterus can lead to pregnancy loss. Health problems such as hormonal imbalance, poorly-controlled diabetes, lupus and other immune system abnormalities, kidney and heart disease, and hypertension can create difficulties in carrying a pregnancy to term. These causes of miscarriage can be evaluated by blood tests and an ultrasound examination (sonogram) of the uterus. Your doctor can evaluate you for these problems. Environment: Another cause of pregnancy loss is an environmental exposure during pregnancy. For example, exposure to drugs, alcohol, or high levels of radiation can lead to miscarriage. Infections can cause miscarriage. The risk of miscarriage may be greater in women who smoke.
Remember, even when repeated miscarriages occur, there is a good chance of successful next pregnancy.
Genetics and osteoporosis
Many things affect your odds of developing osteoporosis. But your genetic and racial background can be significant risk factors. Osteoporosis does run in families, so if your family has a history of the condition, it is even more important for you to take steps now to have healthy bones throughout your life.
Scientists Narrowing in on Genetic Testing
Scientists are steadily gaining an understanding of which genes affect bones. A day will come when genetic testing will be able to rank an individual’s risk for osteoporosis. It may not be too far in the future. But, even if you knew that your risk of osteoporosis is low, it is still worth it to keep up activities that promote bone health -- such as exercise and eating calcium-rich foods -- because those things often reduce the risks of other diseases, as well.
Does Race Affect the Risk of Osteoporosis?
Although there are racial differences, the disease occurs in all racial groups.
However, generally, whites have a greater risk of bone fracture than Asians and Asians have a greater risk than blacks.
Making racial comparisons is complicated because people of different races generally differ from each other by more than just their genes. Besides the genetic differences, they may have very different diet, lifestyle, and environmental factors. For this reason, studies that investigate the effects of diet and exercise on bone health must compare people of similar racial makeup for the study conclusions to be valid.
The poor logic of this statement was well illustrated by a study that was part of a large study conducted by T. Colin Campbell and colleagues from Cornell University. His group measured the bone mineral density (BMD) of morethan 800 Chinese women from five different counties in China. They found that the women with the greatest BMD resided in a pastoral district where milk was a common part of the diet. Their milk consumption resulted in significantly greater consumption of calcium, phosphorus, and protein. So, with all things being genetically similar, increased intake of these nutrients translated into better bones.
The Bottom Line
Regardless of your family and genetic risk, it is still beneficial to to maintain healthy behaviors that are known to reduce the risk of osteoporosis, such as lifting weights and eating adequate amounts of calcium-rich foods.
Scientists Narrowing in on Genetic Testing
Scientists are steadily gaining an understanding of which genes affect bones. A day will come when genetic testing will be able to rank an individual’s risk for osteoporosis. It may not be too far in the future. But, even if you knew that your risk of osteoporosis is low, it is still worth it to keep up activities that promote bone health -- such as exercise and eating calcium-rich foods -- because those things often reduce the risks of other diseases, as well.
Does Race Affect the Risk of Osteoporosis?
Although there are racial differences, the disease occurs in all racial groups.
However, generally, whites have a greater risk of bone fracture than Asians and Asians have a greater risk than blacks.
Making racial comparisons is complicated because people of different races generally differ from each other by more than just their genes. Besides the genetic differences, they may have very different diet, lifestyle, and environmental factors. For this reason, studies that investigate the effects of diet and exercise on bone health must compare people of similar racial makeup for the study conclusions to be valid.
The poor logic of this statement was well illustrated by a study that was part of a large study conducted by T. Colin Campbell and colleagues from Cornell University. His group measured the bone mineral density (BMD) of morethan 800 Chinese women from five different counties in China. They found that the women with the greatest BMD resided in a pastoral district where milk was a common part of the diet. Their milk consumption resulted in significantly greater consumption of calcium, phosphorus, and protein. So, with all things being genetically similar, increased intake of these nutrients translated into better bones.
The Bottom Line
Regardless of your family and genetic risk, it is still beneficial to to maintain healthy behaviors that are known to reduce the risk of osteoporosis, such as lifting weights and eating adequate amounts of calcium-rich foods.
Genetics and breast cancer
Nearly 200,000 American women are diagnosed with breast cancer each year, and between 5 percent to 10 percent of them have a heritable form of the disease. Geneticists have identified mutations on the BRCA1 and BRCA2 genes that predispose a person to develop breast cancer. Women with alterations on these genes have up to an 85% greater lifetime chance of developing breast cancer than women with unaffected genes. The mutations are also associated with an increased risk of ovarian cancer. People of Ashkenazi Jewish descent have a higher incidence of inheriting these mutations than those in other ethnic groups. Although breast cancer in men is far less common, a mutated BRCA2 gene, in particular, increases their possibility of having the disease. Furthermore, people with altered BRCA1 and BRCA2 genes have a slightly greater risk to develop cancer of the pancreas, stomach, or prostate than people without the mutations.
Genetic testing for the mutated BRCA1 or BRCA2 genes should be considered by those who have had ovarian cancer at any age or breast cancer before they were 50 or have relatives meeting the same parameters; people with family histories of two or more cases of breast and/or ovarian cancer; and males with breast cancer. The preferred procedure is to first test a family member who has had breast or ovarian cancer. If this is not possible, then an at-risk family member can be tested directly.
A positive result indicates that the patient is at higher risk for cancer. However, it cannot tell whether cancer will actually occur, or when, or how aggressively. That is why it is important to review test results with an experienced genetic counselor. Some of the options that can be considered in discussion with a genetic counselor include:
Careful monitoring: Increased frequency of mammograms and clinical breast exams can help catch breast cancer at an earlier and more treatable stage. Most importantly, is to begin screening at-risk persons at an earlier age than is recommended for the population at large.
For ovarian cancer, monitoring through transvaginal ultrasound, CA-125 blood tests, and clinical exams can sometimes find cancer at an early stage, but it is not clear if these methods can reduce the risk of death from ovarian cancer.
Prophyltactic Surgery: Removal of the breasts (mastectomy), ovaries and fallopian tubes (salpingo-oophorectomy), or both the breasts and the ovaries can reduce (but not eliminate) the risks of developing breast or ovarian cancer. A Salpingo- oophorectomy may be recommended for women past childbearing age, as ovarian cancer is difficult to detect in its early stages. The younger the patient with altered genes is when she has a preventive mastectomy, the greater the potential benefit. Surgery is a particularly complex and highly personal option to consider and is best made by the patient with information by her physician and her genetic counselor and the support of her family.
Chemoprevention: In general, tamoxifen has been found to be effective in reducing the risk of developing cancer and in reducing the chance that cancer will reoccur in women at increased risk for developing the disease.
Genetic testing for the mutated BRCA1 or BRCA2 genes should be considered by those who have had ovarian cancer at any age or breast cancer before they were 50 or have relatives meeting the same parameters; people with family histories of two or more cases of breast and/or ovarian cancer; and males with breast cancer. The preferred procedure is to first test a family member who has had breast or ovarian cancer. If this is not possible, then an at-risk family member can be tested directly.
A positive result indicates that the patient is at higher risk for cancer. However, it cannot tell whether cancer will actually occur, or when, or how aggressively. That is why it is important to review test results with an experienced genetic counselor. Some of the options that can be considered in discussion with a genetic counselor include:
Careful monitoring: Increased frequency of mammograms and clinical breast exams can help catch breast cancer at an earlier and more treatable stage. Most importantly, is to begin screening at-risk persons at an earlier age than is recommended for the population at large.
For ovarian cancer, monitoring through transvaginal ultrasound, CA-125 blood tests, and clinical exams can sometimes find cancer at an early stage, but it is not clear if these methods can reduce the risk of death from ovarian cancer.
Prophyltactic Surgery: Removal of the breasts (mastectomy), ovaries and fallopian tubes (salpingo-oophorectomy), or both the breasts and the ovaries can reduce (but not eliminate) the risks of developing breast or ovarian cancer. A Salpingo- oophorectomy may be recommended for women past childbearing age, as ovarian cancer is difficult to detect in its early stages. The younger the patient with altered genes is when she has a preventive mastectomy, the greater the potential benefit. Surgery is a particularly complex and highly personal option to consider and is best made by the patient with information by her physician and her genetic counselor and the support of her family.
Chemoprevention: In general, tamoxifen has been found to be effective in reducing the risk of developing cancer and in reducing the chance that cancer will reoccur in women at increased risk for developing the disease.
Genetics and blood clotting disorders
Thrombosis is the presence or formation of a thrombus - a blood clot that forms within a vein or an artery. The condition arising from clots developing in the body’s deep veins (typically the leg and pelvic regions) is known as Deep Vein Thrombosis, or DVT. Sometimes the blood clot dissolves on its own, with no harm. But a clot that prohibits adequate amounts of blood to pass through a vein can result in necrosis, or tissue death. Even worse, the clot could break off, travel through the blood stream, and lodge itself in an artery in the lungs, causing a life-threatening condition known as pulmonary embolism (PE).
There are many reasons why these abnormal clots form. When a person’s ability to move about is limited, whether due to a long airplane flight (“Economy Class Syndrome”) or confinement to a bed after surgery, blood can pool and form clots. The use of oral contraceptives or estrogen therapy can also cause clotting.
Genetics can have a role in the development of thrombosis. There are several known mutations (heritable following an autosomal dominant pattern) that interfere with the body’s blood clotting process. The most common is the Factor V Leiden mutation, a resistance to Protein C, a protein that keeps the formation of blood clots in check. Present in 5 – 7 percent of the general population, the Factor V Leiden mutation is present in 20 percent of more of all people with thrombosis. Less frequently occurring mutations include Prothrombin II, Protein C deficiency; Protein S deficiency; Antithrombin III deficiency; Plasminogen deficiency; and Heparin cofactor II deficiency. It is possible for a person to have more than one of these mutations. When the Prothrombin II mutation is present with the Factor V Leiden mutation, the risk for DVT or PE is increased substantially.
The genetic test for Factor V Leiden and Prothrombin II is a DNA analysis taken from a sample of blood or cheek cells. There are also panel tests that screen for several of the mutations described above. Testing should be considered for: people who experience a thrombotic event—such as a DVT or PE— before age 50 or have multiple thrombotic events; those with a family history of thrombotic events, even if they themselves are not experiencing symptoms; and people with a first-degree relative who is known to have one of the thrombosis mutations.
A person who has had a thrombotic event and, after testing, is found to have the Factor V Leiden mutation or any of the other thrombilia-related mutations, is usually advised to take anticoagulant medication, such as Coumadin. The period of time in which the medication is taken will depend upon the specific circumstances surrounding the thrombotic event.
Additionally, affected persons as well as family members who have a mutation but no history or indication of clotting disorders would be advised to take preventive steps, such as not smoking, not taking birth control pills, wearing support hose and stretching during long airplane flights, and alerting their physicians if they are scheduled for surgery or are immobilized for a long period of time.
There are many reasons why these abnormal clots form. When a person’s ability to move about is limited, whether due to a long airplane flight (“Economy Class Syndrome”) or confinement to a bed after surgery, blood can pool and form clots. The use of oral contraceptives or estrogen therapy can also cause clotting.
Genetics can have a role in the development of thrombosis. There are several known mutations (heritable following an autosomal dominant pattern) that interfere with the body’s blood clotting process. The most common is the Factor V Leiden mutation, a resistance to Protein C, a protein that keeps the formation of blood clots in check. Present in 5 – 7 percent of the general population, the Factor V Leiden mutation is present in 20 percent of more of all people with thrombosis. Less frequently occurring mutations include Prothrombin II, Protein C deficiency; Protein S deficiency; Antithrombin III deficiency; Plasminogen deficiency; and Heparin cofactor II deficiency. It is possible for a person to have more than one of these mutations. When the Prothrombin II mutation is present with the Factor V Leiden mutation, the risk for DVT or PE is increased substantially.
The genetic test for Factor V Leiden and Prothrombin II is a DNA analysis taken from a sample of blood or cheek cells. There are also panel tests that screen for several of the mutations described above. Testing should be considered for: people who experience a thrombotic event—such as a DVT or PE— before age 50 or have multiple thrombotic events; those with a family history of thrombotic events, even if they themselves are not experiencing symptoms; and people with a first-degree relative who is known to have one of the thrombosis mutations.
A person who has had a thrombotic event and, after testing, is found to have the Factor V Leiden mutation or any of the other thrombilia-related mutations, is usually advised to take anticoagulant medication, such as Coumadin. The period of time in which the medication is taken will depend upon the specific circumstances surrounding the thrombotic event.
Additionally, affected persons as well as family members who have a mutation but no history or indication of clotting disorders would be advised to take preventive steps, such as not smoking, not taking birth control pills, wearing support hose and stretching during long airplane flights, and alerting their physicians if they are scheduled for surgery or are immobilized for a long period of time.
Genetics and epilepsy
What is genetics?
Genetics is the study of genes, which are the basic units of heredity. Human beings have many thousands of genes. Each of these genes influences certain traits such as hair color, eye color, blood type, and many other characteristics. People are different with regard to these traits because their genes are different. Children look similar to one or both parents, for example, or have traits similar to their grandparents and other relatives, because of certain genes which have been passed or inherited from one generation to the next.
I thought we inherited different traits because of our chromosomes. What is the difference between genes and chromosomes?
An easy way of thinking about a chromosome is as a "package" filled with many genes.
The human body is made up of millions of cells. There are many different types of cells such as brain cells, muscle cells, and skin cells. Most cells in the body contain chromosomes. Each of these cells holds 23 pairs of chromosomes (46 total). One member of each pair is inherited from the mother and the other from the father. Genes are located on chromosomes and, thus, are passed from both parents to the child.
Genes, in turn, are made up of a substance called DNA (deoxyribonucleic acid). Genes give the cell instructions to make proteins, which are necessary for the body to do all the things that it does.
That sounds so complicated. What if something goes wrong?
It is complicated. Genes can be altered in many ways. Changes in the DNA that cause a protein to not work normally or stop working altogether are called "mutations." Environmental factors can also affect the way cells work.
Some mutations may result in disease and some of these conditions can be passed on to future generations.
Is epilepsy inherited?
Some types of epilepsy are. Epilepsy is not a single disorder, but a collection of many disorders that all have in common the tendency to cause a person to have seizures. When individual characteristics are caused by single genes, they are called "simple" genetic traits. Only a few rare types of epilepsy are caused by alterations in single genes. Most seem to be caused by a complex interaction among multiple genes and environmental influences.
I have epilepsy. Is my child likely to have seizures, too?
Certain types of epilepsy do seem to run in families. Some studies have shown that the risk of epilepsy in brothers, sisters and children of people with seizure disorders ranges from 4-8% (that is, between about 1 in 25 and about 1 in 12). The risk in the general population is about 1-2% (between 1 in 100 and 1 in 50). However, it's important to remember that although the risk is higher than in the general population, most people with epilepsy do not have any relatives with seizures, and the great majority of parents with epilepsy do not have children with epilepsy.
Which types of epilepsy are most likely to be inherited?
People who have a generalized epilepsy (one where the EEG pattern shows both sides of the brain involved at the beginning of a seizure) seem somewhat more likely to have other family members with seizures than those with a localization-related epilepsy (also called partial or focal, where the EEG pattern shows seizures beginning in a single area of the brain). Parents with a history of generalized absence (childhood petit mal) seizures are more likely to have children with the same condition than those with other generalized seizures or focal seizures.
What are some of the other factors that seem to be involved in inheritance?
1) Age when epilepsy begins -- Children of people whose seizures started early in life (for example, before 20 years of age) have a greater risk of developing epilepsy than children of people whose seizures started later in life.
2) Mothers and fathers with epilepsy -- Studies have shown that the risk of epilepsy is about twice as high in children of women with epilepsy than in children of men with epilepsy. Research has shown that this is not related to pregnancy or birth complications, maternal seizures during pregnancy or maternal antiepileptic drugs during pregnancy. More research is needed to explain this difference.
3) Cause of epilepsy -- The risk for developing epilepsy does not seem to be increased, compared with the general population, in relatives of people who have epilepsy caused by serious brain injury that occurs after birth due to conditions such as strokes, brain tumors, severe head trauma or brain infections.
Can epilepsy skip a generation?
Yes, it can. As discussed earlier, most types of epilepsy are caused by multiple genes and environmental influences. Not everyone who carries genes that make him or her more likely to develop epilepsy will, in fact, do so. Therefore, even if the genes are passed on, not every generation in a family will have seizures.
How can I find out what the risk is for my baby to inherit my type of epilepsy?
Ask your physician to refer you for genetic counseling. Specially trained physicians or nurses, genetic counselors, and other health care professionals can help you study your medical history, find out facts about your family history and, if possible, calculate the risk for you and your baby. They may recommend certain laboratory tests to get more information.
It's important to remember that although there is a lot of evidence that genes play an important role in causing epilepsy, exactly which genes are involved has not been identified for most people who have seizures. Studying the families of people who have epilepsy will help increase our knowledge and perhaps, in the future, will lead to new treatments for epilepsy or even measures to prevent epilepsy.
If more than one member of your family has a history of epilepsy or seizures, please consider participating in the Foundation's Gene Discovery Project. Completion of a confidential online questionnaire by you or a family member will allow scientists to determine whether the pattern of epilepsy found in your family could be valuable for further study. Your participation may help lead to exciting new discoveries about genetics and epilepsy.
Genetics is the study of genes, which are the basic units of heredity. Human beings have many thousands of genes. Each of these genes influences certain traits such as hair color, eye color, blood type, and many other characteristics. People are different with regard to these traits because their genes are different. Children look similar to one or both parents, for example, or have traits similar to their grandparents and other relatives, because of certain genes which have been passed or inherited from one generation to the next.
I thought we inherited different traits because of our chromosomes. What is the difference between genes and chromosomes?
An easy way of thinking about a chromosome is as a "package" filled with many genes.
The human body is made up of millions of cells. There are many different types of cells such as brain cells, muscle cells, and skin cells. Most cells in the body contain chromosomes. Each of these cells holds 23 pairs of chromosomes (46 total). One member of each pair is inherited from the mother and the other from the father. Genes are located on chromosomes and, thus, are passed from both parents to the child.
Genes, in turn, are made up of a substance called DNA (deoxyribonucleic acid). Genes give the cell instructions to make proteins, which are necessary for the body to do all the things that it does.
That sounds so complicated. What if something goes wrong?
It is complicated. Genes can be altered in many ways. Changes in the DNA that cause a protein to not work normally or stop working altogether are called "mutations." Environmental factors can also affect the way cells work.
Some mutations may result in disease and some of these conditions can be passed on to future generations.
Is epilepsy inherited?
Some types of epilepsy are. Epilepsy is not a single disorder, but a collection of many disorders that all have in common the tendency to cause a person to have seizures. When individual characteristics are caused by single genes, they are called "simple" genetic traits. Only a few rare types of epilepsy are caused by alterations in single genes. Most seem to be caused by a complex interaction among multiple genes and environmental influences.
I have epilepsy. Is my child likely to have seizures, too?
Certain types of epilepsy do seem to run in families. Some studies have shown that the risk of epilepsy in brothers, sisters and children of people with seizure disorders ranges from 4-8% (that is, between about 1 in 25 and about 1 in 12). The risk in the general population is about 1-2% (between 1 in 100 and 1 in 50). However, it's important to remember that although the risk is higher than in the general population, most people with epilepsy do not have any relatives with seizures, and the great majority of parents with epilepsy do not have children with epilepsy.
Which types of epilepsy are most likely to be inherited?
People who have a generalized epilepsy (one where the EEG pattern shows both sides of the brain involved at the beginning of a seizure) seem somewhat more likely to have other family members with seizures than those with a localization-related epilepsy (also called partial or focal, where the EEG pattern shows seizures beginning in a single area of the brain). Parents with a history of generalized absence (childhood petit mal) seizures are more likely to have children with the same condition than those with other generalized seizures or focal seizures.
What are some of the other factors that seem to be involved in inheritance?
1) Age when epilepsy begins -- Children of people whose seizures started early in life (for example, before 20 years of age) have a greater risk of developing epilepsy than children of people whose seizures started later in life.
2) Mothers and fathers with epilepsy -- Studies have shown that the risk of epilepsy is about twice as high in children of women with epilepsy than in children of men with epilepsy. Research has shown that this is not related to pregnancy or birth complications, maternal seizures during pregnancy or maternal antiepileptic drugs during pregnancy. More research is needed to explain this difference.
3) Cause of epilepsy -- The risk for developing epilepsy does not seem to be increased, compared with the general population, in relatives of people who have epilepsy caused by serious brain injury that occurs after birth due to conditions such as strokes, brain tumors, severe head trauma or brain infections.
Can epilepsy skip a generation?
Yes, it can. As discussed earlier, most types of epilepsy are caused by multiple genes and environmental influences. Not everyone who carries genes that make him or her more likely to develop epilepsy will, in fact, do so. Therefore, even if the genes are passed on, not every generation in a family will have seizures.
How can I find out what the risk is for my baby to inherit my type of epilepsy?
Ask your physician to refer you for genetic counseling. Specially trained physicians or nurses, genetic counselors, and other health care professionals can help you study your medical history, find out facts about your family history and, if possible, calculate the risk for you and your baby. They may recommend certain laboratory tests to get more information.
It's important to remember that although there is a lot of evidence that genes play an important role in causing epilepsy, exactly which genes are involved has not been identified for most people who have seizures. Studying the families of people who have epilepsy will help increase our knowledge and perhaps, in the future, will lead to new treatments for epilepsy or even measures to prevent epilepsy.
If more than one member of your family has a history of epilepsy or seizures, please consider participating in the Foundation's Gene Discovery Project. Completion of a confidential online questionnaire by you or a family member will allow scientists to determine whether the pattern of epilepsy found in your family could be valuable for further study. Your participation may help lead to exciting new discoveries about genetics and epilepsy.
Genetics and alcoholism
The contribution of genetics to an understanding alcoholism and other diseases having addictive behavior has been wrought with controversy for the past two hundred years. Because this is a politically and socially charged issue, there has been much debate regarding the true genetic contribution to alcoholism. Traditional medicine states that disease can be attributed to certain environmental conditions, specific gene alleles inherited from the parents or some combination of both of these factors. Most estimates of the contribution of genetics to alcoholism put the contribution of genetics about equal to that of the environment. Thus, the contribution of genetics to the disease is said to be about 50%. It should be noted, however, that various researchers have put this contribution as low as 10% or as high as 70%. The argument for heredity having a relatively strong influence on this disease rests primarily in studies involving families, adoptees and twins.
There is strong evidence that alcoholism runs in families. Most research studies in this area have demonstrated that about one fourth of the sons of alcoholics become alcoholics themselves. Daughters of alcoholics develop this disease about 5% of the time. While the estimates for the rate of alcoholism vary greatly for the general population, these rates are usually higher. In fact, the most consistent risk factor for developing alcoholism is a strong family history. Despite this data that shows a familial relationship for alcoholism, it could be argued that it is a learned behavior. Evidence from adoption studies further supports the contention of a genetic basis for alcoholism.
The use of adoptees is a common method to attempt to separate the effects of the environment from genetics. The rationale behind this method is that if biological children of alcoholic parents develop the disease at a greater rate than the general population when they reside with parents who do not have the disease there must be a strong genetic component to the disease. Most studies in this area have concluded that despite residing with adoptive, nonalcoholic parents, children who had alcoholic biological parents were at high risk for alcoholism. These rates reported were similar to that of children who grew up in the homes of their alcoholic biological parents. These studies strongly suggest a hereditary basis for alcoholism. This contention is further supported by twin studies which show that a second identical twin is much more likely to develop the disease if the first one developed it. Because identical twins have the same genetic instructions, this finding is consistent with the contention of a genetic component to alcoholism.
Other research models also support the assertion that heredity plays an important role in alcoholism. Researchers have turned to the science of molecular biology in an attempt to decipher this complex problem. One of the more compelling findings was that genetically engineered mice that had lacked a specific dopamine receptor gene in the brain were less likely to prefer alcohol and have a sensitivity to it than siblings that had the receptor. The results indicated that taking away the receptor decreased alcohol consumption by 50%. Although extensive testing needs to be completed, it is possible that mutations of these receptors in the brain may contribute to alcoholism in humans. If this avenue of research proves to be fruitful, it may be possible to treat alcoholism in the distant future through manipulation of this gene.
It is likely that alcoholism results from a combination of both genetic and environmental factors. In fact, most researchers working in this area believe that it is unlikely that science will determine the alcoholism gene. Rather, it is likely that the interaction of multiple genes contributes to the development of alcoholism. It should also be noted that those individuals with this array of genes are not predetermined to be alcoholics. While there is tremendous evidence that genes can exert influence over behavior, there is little support for the contention that they cause it. Thus, environment still plays a vital role in the development of this disease. While the exact alleles and their specific contribution to the development of alcoholism cannot be concluded with any certainty, it is known that genetics plays a role in the development of this disease, the actual mechanisms on how this happens has yet to be discovered by science.
There is strong evidence that alcoholism runs in families. Most research studies in this area have demonstrated that about one fourth of the sons of alcoholics become alcoholics themselves. Daughters of alcoholics develop this disease about 5% of the time. While the estimates for the rate of alcoholism vary greatly for the general population, these rates are usually higher. In fact, the most consistent risk factor for developing alcoholism is a strong family history. Despite this data that shows a familial relationship for alcoholism, it could be argued that it is a learned behavior. Evidence from adoption studies further supports the contention of a genetic basis for alcoholism.
The use of adoptees is a common method to attempt to separate the effects of the environment from genetics. The rationale behind this method is that if biological children of alcoholic parents develop the disease at a greater rate than the general population when they reside with parents who do not have the disease there must be a strong genetic component to the disease. Most studies in this area have concluded that despite residing with adoptive, nonalcoholic parents, children who had alcoholic biological parents were at high risk for alcoholism. These rates reported were similar to that of children who grew up in the homes of their alcoholic biological parents. These studies strongly suggest a hereditary basis for alcoholism. This contention is further supported by twin studies which show that a second identical twin is much more likely to develop the disease if the first one developed it. Because identical twins have the same genetic instructions, this finding is consistent with the contention of a genetic component to alcoholism.
Other research models also support the assertion that heredity plays an important role in alcoholism. Researchers have turned to the science of molecular biology in an attempt to decipher this complex problem. One of the more compelling findings was that genetically engineered mice that had lacked a specific dopamine receptor gene in the brain were less likely to prefer alcohol and have a sensitivity to it than siblings that had the receptor. The results indicated that taking away the receptor decreased alcohol consumption by 50%. Although extensive testing needs to be completed, it is possible that mutations of these receptors in the brain may contribute to alcoholism in humans. If this avenue of research proves to be fruitful, it may be possible to treat alcoholism in the distant future through manipulation of this gene.
It is likely that alcoholism results from a combination of both genetic and environmental factors. In fact, most researchers working in this area believe that it is unlikely that science will determine the alcoholism gene. Rather, it is likely that the interaction of multiple genes contributes to the development of alcoholism. It should also be noted that those individuals with this array of genes are not predetermined to be alcoholics. While there is tremendous evidence that genes can exert influence over behavior, there is little support for the contention that they cause it. Thus, environment still plays a vital role in the development of this disease. While the exact alleles and their specific contribution to the development of alcoholism cannot be concluded with any certainty, it is known that genetics plays a role in the development of this disease, the actual mechanisms on how this happens has yet to be discovered by science.
Genetics and depression
It has long been known that depressive illnesses can run in families, but until fairly recently it was not fully known whether people inherited a susceptibility to these illnesses or if something else such as the environment was the true culprit. Those who research depression have been able to determine that to some extent depressive illnesses can be inherited. What appears to be inherited is a vulnerability to depression. This means that if we have close relatives who have clinical depression, we may inherit a tendency to develop the illness. It does not mean that we are destined to become depressed.
Genes that we inherit from our parents determine many things about us such as our gender and the color of our eyes and hair. Our genes also determine which illnesses we may be vulnerable to at some point in our lives. Every cell in the human body contains somewhere between 50,000 and 100,000 genes. They are all made up of something called deoxyribonucleic acid, or DNA. Genes are located on chromosomes within the nucleus of each cell. All of the cells in the body, except sex cells, contain 46 chromosomes, and genes are typically located in a specific place on a particular chromosome. Except for identical twins, no two people in the world have the exact same genetic makeup.
Research on the heredity of depression within families shows that some individuals are more likely to develop the illness than others. If you have a parent or sibling that has had major depression, you may be 1.5 to 3 times more likely to develop the condition than those who do not have a close relative with the condition. You would also have a higher chance of developing bipolar disorder. Because close relatives of those with clinical depression have such a vulnerability to developing the condition themselves strongly suggests that it can be an inherited illness.
Bipolar disorder has a strong genetic influence. Of those with bipolar disorder, approximately 50% of them have a parent with a history of clinical depression. When a mother or father has bipolar disorder, their child will have a 25% chance of developing some type of clinical depression. If both parents have bipolar disorder, the chance of their child also developing bipolar disorder is between 50% and 75%. Brothers and sisters of those with bipolar disorder may be 8 to 18 times more likely to develop bipolar disorder, and 2 to 10 times more likely to develop major depressive disorder than others with no such siblings.
Twin Studies
Much of what we know about the genetic influence of clinical depression is based upon research that has been done with identical twins. Identical twins are very helpful to researchers since they both have the exact same genetic code. It has been found that when one identical twin becomes depressed the other will also develop clinical depression approximately 76% of the time. When identical twins are raised apart from each other, they will both become depressed about 67% of the time. Because both twins become depressed at such a high rate, the implication is that there is a strong genetic influence. If it happened that when one twin becomes clinically depressed the other always develops depression, then clinical depression would likely be entirely genetic. However because the rate of both identical twins developing depression is not closer to 100% this tells us that there are other things that influence a person's vulnerability to depression. These may include environmental factors such as childhood experiences, current stressors, traumatic events, exposure to substances, medical illnesses, etc.
Research has also been done with fraternal twins. Unlike identical twins who have the same genetic code, these siblings share only about 50% of their genetic makeup and do not necessarily look alike. Studies have shown that when one fraternal twin becomes depressed, the other also develops depression about 19% of the time. This is still a higher rate of depression when compared to overall rates for the general public, again pointing towards a genetic influence in the development of clinical depression.
A Gene for Depression?
Research on the genetic causes of clinical depression has attempted to identify one or more specific genes that may lead to the development of a depressive illness. Although there have been a number of studies that appear to name a particular gene as the culprit there has been little consistency among their results. However, the outcome of some research has suggested that there may be specific genes that cause clinical depression to develop within certain families and not in others.
At this time there is much that we do not know about how genes may predispose a person to a depressive illness. Research has yet to identify a clear link between a specific gene and a vulnerability to depression in everyone. Rather than the possibility of only a single gene being responsible for the development of clinical depression, it appears to be more likely that a number of genes acting together may cause a person to become vulnerable to depression.
Just because a person inherits a gene that predisposes him or her to a depressive illness, it does not mean that he or she is destined to develop major depression or bipolar disorder. It is believed that a genetic influence is only partially responsible for causing depression. Other factors may also play a role.
Genes that we inherit from our parents determine many things about us such as our gender and the color of our eyes and hair. Our genes also determine which illnesses we may be vulnerable to at some point in our lives. Every cell in the human body contains somewhere between 50,000 and 100,000 genes. They are all made up of something called deoxyribonucleic acid, or DNA. Genes are located on chromosomes within the nucleus of each cell. All of the cells in the body, except sex cells, contain 46 chromosomes, and genes are typically located in a specific place on a particular chromosome. Except for identical twins, no two people in the world have the exact same genetic makeup.
Research on the heredity of depression within families shows that some individuals are more likely to develop the illness than others. If you have a parent or sibling that has had major depression, you may be 1.5 to 3 times more likely to develop the condition than those who do not have a close relative with the condition. You would also have a higher chance of developing bipolar disorder. Because close relatives of those with clinical depression have such a vulnerability to developing the condition themselves strongly suggests that it can be an inherited illness.
Bipolar disorder has a strong genetic influence. Of those with bipolar disorder, approximately 50% of them have a parent with a history of clinical depression. When a mother or father has bipolar disorder, their child will have a 25% chance of developing some type of clinical depression. If both parents have bipolar disorder, the chance of their child also developing bipolar disorder is between 50% and 75%. Brothers and sisters of those with bipolar disorder may be 8 to 18 times more likely to develop bipolar disorder, and 2 to 10 times more likely to develop major depressive disorder than others with no such siblings.
Twin Studies
Much of what we know about the genetic influence of clinical depression is based upon research that has been done with identical twins. Identical twins are very helpful to researchers since they both have the exact same genetic code. It has been found that when one identical twin becomes depressed the other will also develop clinical depression approximately 76% of the time. When identical twins are raised apart from each other, they will both become depressed about 67% of the time. Because both twins become depressed at such a high rate, the implication is that there is a strong genetic influence. If it happened that when one twin becomes clinically depressed the other always develops depression, then clinical depression would likely be entirely genetic. However because the rate of both identical twins developing depression is not closer to 100% this tells us that there are other things that influence a person's vulnerability to depression. These may include environmental factors such as childhood experiences, current stressors, traumatic events, exposure to substances, medical illnesses, etc.
Research has also been done with fraternal twins. Unlike identical twins who have the same genetic code, these siblings share only about 50% of their genetic makeup and do not necessarily look alike. Studies have shown that when one fraternal twin becomes depressed, the other also develops depression about 19% of the time. This is still a higher rate of depression when compared to overall rates for the general public, again pointing towards a genetic influence in the development of clinical depression.
A Gene for Depression?
Research on the genetic causes of clinical depression has attempted to identify one or more specific genes that may lead to the development of a depressive illness. Although there have been a number of studies that appear to name a particular gene as the culprit there has been little consistency among their results. However, the outcome of some research has suggested that there may be specific genes that cause clinical depression to develop within certain families and not in others.
At this time there is much that we do not know about how genes may predispose a person to a depressive illness. Research has yet to identify a clear link between a specific gene and a vulnerability to depression in everyone. Rather than the possibility of only a single gene being responsible for the development of clinical depression, it appears to be more likely that a number of genes acting together may cause a person to become vulnerable to depression.
Just because a person inherits a gene that predisposes him or her to a depressive illness, it does not mean that he or she is destined to develop major depression or bipolar disorder. It is believed that a genetic influence is only partially responsible for causing depression. Other factors may also play a role.
Genetics and Alzheimers disease
The genetic factors associated with Alzheimer's disease can be summarised as follows:
There is no single gene for Alzheimer's disease.
Genetic factors are responsible for the disease in a small number of families.
In the wider community there is a genetic component to the disease, but inherited factors alone do not explain why some people develop it while others do not.
Genes and Inheritance
The basic material of inheritance, DNA, is passed on in the form of genes, and delivered in packages called chromosomes, which are long chains of genetic instructions. We can think of DNA as being the letters of the alphabet, genes as the words they make up and chromosomes the sentences which convey meaning.
We each inherit 23 pairs of chromosomes, one half of each pair from our mother and the other from our father. There are literally millions of combinations of genes we can inherit, and the effect of each gene is not yet known, although scientists worldwide are rapidly expanding our knowledge.
Alzheimer's disease genes
Alzheimer s disease runs in a small number of families, where up to half of the family members are at risk.
Alzheimer's disease is common amongst elderly people, so having one close relative with the disease is not evidence of a family link. Even where there are two close relatives with the disease it is likely to have occurred by chance.
Early onset
If you have three or more close relatives who developed Alzheimer s disease at an early age, then your doctor will be able to counsel you about genetic testing for familial Alzheimer s disease and refer you to a geneticist where appropriate.
About 15 families in the world have a genetic fault on chromosome 21 in a gene called amyloid precursor protein, or APP, which affects production of a protein called amyloid. This protein has been associated with Alzheimer's when it builds up in the brain.
A larger number of families carry a fault on chromosome 14 ('presenilin-1') which could be responsible for the majority of the early onset cases of familial Alzheimer's, sometimes below the age of 40.
A smaller group of families (mainly in the United States) has a fault on chromosome 1, which has been named presenilin-2 .
All these genetic faults are associated with early onset of Alzheimer's disease, usually between ages of 35 and 60.
On average half the children of someone with one of these genetic defects inherits it, and probably all those who inherit the gene develop the disease. Those who do not inherit cannot pass on the genetic faults. They do not skip a generation.
Later onset
Most cases of Alzheimer's develop later in life. Below the age of 65 the risk is approximately 1 in 1000. Over the age of 65 it affects one person in 50. The risk rises to one person on five by the age of 80
There is also a genetic link with many later cases, weaker than the link described above, but not confined to a few families.
The link is with a protein called apolipo-protein E (ApoE), which we all have in the blood and the brain. It comes in three forms, known as ApoE2, ApoE3 and ApoE4, all found in the general population. We each have two copies of the gene, which may be the same as each other or different.
ApoE4 is associated with higher risk of Alzheimer's disease. About a quarter of the population inherit one copy of the ApoE4 gene and this increases the risk of developing Alzheimer's disease by up to four times.
Two per cent of the population get a double dose of the ApoE4 gene, one from each parent. The risk of Alzheimer's disease is increased by 16 times in this group, but it is still not inevitable that the disease will develop.
Sixty per cent of the population have a double dose of the ApoE3 gene and are at average risk . About half develop Alzheimer's by their late 80s.
ApoE2 is the gene least associated with Alzheimer's disease, but only one in six people carries it. People who have one ApoE2 gene and one ApoE3 gene (11 per cent of the population) have to live into their late 90s before their risk of Alzheimer's diseases reaches 50 per cent.
One in 200 people inherit two copies of the ApoE2 gene and are at very low risk of Alzheimer's disease.
The ApoE risk is very different from familial Alzheimer's. ApoE4 increases the chances of the disease, but does not make it certain. Some other factor, not yet understood, must also contribute.
It is likely that there are many other genes that might contribute to increasing risk of Alzheimer's disease. Genes linked by recent research to late onset Alzheimer's disease include certain apolipoprotein receptors, the serotonin transporter, a small change in the presenilin-1 gene, butyrylcholinesterase K, the HLA gene and a region on chromosome 12.
Much more research remains to be done before geneticists can be sure that these genes are risk factors for Alzheimer's disease and in any case it is quite likely that the degree of risk involved will be individually quite small.
What are the pros and cons of genetic testing?
A genetic test for Alzheimer's could:
identify people who might benefit from new drugs to delay symptoms in the early stages of Alzheimer's disease
help genetic researchers understand the disease better and so lead to better treatment.
help some people plan for the future
However, population-wide screening would create problems for individuals and for society.
A genetic defect cannot be repaired, and effective treatment is not yet generally available, so a test could raise anxiety without offering a clear course of action.
A genetic test for ApoE4 cannot accurately predict who will develop the disease testing positive does not mean you will get it; testing negative does not guarantee you will not.
People who test positive could face discrimination which could damage their ability to buy a house, obtain insurance or plan financially for their old age.
If you have three or more close family members with early onset of dementia, you may want to be referred to a specialist centre for genetic testing. Your GP will counsel you and help to assess your risk. You should discuss the issues carefully before making a decision.
There is no single gene for Alzheimer's disease.
Genetic factors are responsible for the disease in a small number of families.
In the wider community there is a genetic component to the disease, but inherited factors alone do not explain why some people develop it while others do not.
Genes and Inheritance
The basic material of inheritance, DNA, is passed on in the form of genes, and delivered in packages called chromosomes, which are long chains of genetic instructions. We can think of DNA as being the letters of the alphabet, genes as the words they make up and chromosomes the sentences which convey meaning.
We each inherit 23 pairs of chromosomes, one half of each pair from our mother and the other from our father. There are literally millions of combinations of genes we can inherit, and the effect of each gene is not yet known, although scientists worldwide are rapidly expanding our knowledge.
Alzheimer's disease genes
Alzheimer s disease runs in a small number of families, where up to half of the family members are at risk.
Alzheimer's disease is common amongst elderly people, so having one close relative with the disease is not evidence of a family link. Even where there are two close relatives with the disease it is likely to have occurred by chance.
Early onset
If you have three or more close relatives who developed Alzheimer s disease at an early age, then your doctor will be able to counsel you about genetic testing for familial Alzheimer s disease and refer you to a geneticist where appropriate.
About 15 families in the world have a genetic fault on chromosome 21 in a gene called amyloid precursor protein, or APP, which affects production of a protein called amyloid. This protein has been associated with Alzheimer's when it builds up in the brain.
A larger number of families carry a fault on chromosome 14 ('presenilin-1') which could be responsible for the majority of the early onset cases of familial Alzheimer's, sometimes below the age of 40.
A smaller group of families (mainly in the United States) has a fault on chromosome 1, which has been named presenilin-2 .
All these genetic faults are associated with early onset of Alzheimer's disease, usually between ages of 35 and 60.
On average half the children of someone with one of these genetic defects inherits it, and probably all those who inherit the gene develop the disease. Those who do not inherit cannot pass on the genetic faults. They do not skip a generation.
Later onset
Most cases of Alzheimer's develop later in life. Below the age of 65 the risk is approximately 1 in 1000. Over the age of 65 it affects one person in 50. The risk rises to one person on five by the age of 80
There is also a genetic link with many later cases, weaker than the link described above, but not confined to a few families.
The link is with a protein called apolipo-protein E (ApoE), which we all have in the blood and the brain. It comes in three forms, known as ApoE2, ApoE3 and ApoE4, all found in the general population. We each have two copies of the gene, which may be the same as each other or different.
ApoE4 is associated with higher risk of Alzheimer's disease. About a quarter of the population inherit one copy of the ApoE4 gene and this increases the risk of developing Alzheimer's disease by up to four times.
Two per cent of the population get a double dose of the ApoE4 gene, one from each parent. The risk of Alzheimer's disease is increased by 16 times in this group, but it is still not inevitable that the disease will develop.
Sixty per cent of the population have a double dose of the ApoE3 gene and are at average risk . About half develop Alzheimer's by their late 80s.
ApoE2 is the gene least associated with Alzheimer's disease, but only one in six people carries it. People who have one ApoE2 gene and one ApoE3 gene (11 per cent of the population) have to live into their late 90s before their risk of Alzheimer's diseases reaches 50 per cent.
One in 200 people inherit two copies of the ApoE2 gene and are at very low risk of Alzheimer's disease.
The ApoE risk is very different from familial Alzheimer's. ApoE4 increases the chances of the disease, but does not make it certain. Some other factor, not yet understood, must also contribute.
It is likely that there are many other genes that might contribute to increasing risk of Alzheimer's disease. Genes linked by recent research to late onset Alzheimer's disease include certain apolipoprotein receptors, the serotonin transporter, a small change in the presenilin-1 gene, butyrylcholinesterase K, the HLA gene and a region on chromosome 12.
Much more research remains to be done before geneticists can be sure that these genes are risk factors for Alzheimer's disease and in any case it is quite likely that the degree of risk involved will be individually quite small.
What are the pros and cons of genetic testing?
A genetic test for Alzheimer's could:
identify people who might benefit from new drugs to delay symptoms in the early stages of Alzheimer's disease
help genetic researchers understand the disease better and so lead to better treatment.
help some people plan for the future
However, population-wide screening would create problems for individuals and for society.
A genetic defect cannot be repaired, and effective treatment is not yet generally available, so a test could raise anxiety without offering a clear course of action.
A genetic test for ApoE4 cannot accurately predict who will develop the disease testing positive does not mean you will get it; testing negative does not guarantee you will not.
People who test positive could face discrimination which could damage their ability to buy a house, obtain insurance or plan financially for their old age.
If you have three or more close family members with early onset of dementia, you may want to be referred to a specialist centre for genetic testing. Your GP will counsel you and help to assess your risk. You should discuss the issues carefully before making a decision.
Genetics and arthritis
Is Arthritis All In The Family?
Why Me? Is It In The Genes?
Why does one person get arthritis as opposed to another person?
Why is a person afflicted with rheumatoid arthritis rather than osteoarthritis?
Why does one person have it much more severely than another person?
The endless "why" questions traverse the minds of people who suffer with arthritis, the people who care about them, the doctors who treat them, and the researchers who strive to find the answers.
Researchers and scientists are focused on what causes arthritis and what predisposes certain people to the disease. Many things have been considered as contributing to the cause of arthritis, including:
viruses
environmental factors
diet
other triggers
These and other factors have all been considered as relating to the cause.
zSB(3,3)
During the past few decades, genetics has become a more prominent area of arthritis research.
HLA-B27 Gene
A major genetic link was discovered back in the 1960's between the gene HLA-B27 and the spondyloarthropathies, a group of diseases affecting the spine and other joints.
90 percent of people with ankylosing spondylitis have the HLA-B27 gene.
7 percent of the general population have the HLA-B27 gene.
Scientists are working hard to find the other genes involved in the spondyloarthropathies and other rheumatic diseases.
During the decade following the discovery of HLA-B27, scientists found an association between carriers of the HLA-DR genes and increased risk of rheumatoid arthritis. It is believed that HLA-DR contributes to autoimmune disease, conditions caused by the immune system fighting the body it is supposed to protect. Other genes also are suspected of being major factors in the evolution of rheumatoid arthritis.
North American Rheumatoid Arthritis Consortium
The North American Rheumatoid Arthritis Consortium (which consists of 12 medical centers across the country, established and sponsored by the Arthritis Foundation, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Institute of Allergy and Infectious Diseases) has been analyzing clinical findings and genetic material from 1,000 pairs of siblings who both have rheumatoid arthritis. By testing 400 different genetic regions, researchers hope to identify specific genes associated with the rheumatoid arthritis.
HLA-DR4 Gene
The HLA-DR4 gene, which has been associated with rheumatoid arthritis, has also shown involvement inLyme disease. Lyme disease is caused by a microorganism which is transmitted to humans via deer ticks. Among the symptoms which can develop from Lyme disease are:
joint pain
inflammation
arthritis
People who have the disease more severely and do not respond well to the antibiotic treatment are more often found to have the HLA-DR4 gene. It has been theorized that once the microorganism moves to the joints, the immune response against it cross reacts with the person's own joint tissue in people who have the HLA-DR4 gene, leading to an autoimmune reaction.
Genetic Mutations
Other research has led to the discovery of a genetic mutation which causes osteoarthritis in some people. Several members of a family who had early onset osteoarthritis were found to have a genetic mutation in type II collagen. The mutation caused premature breakdown of joint cartilage in the affected family members. Since this discovery other genetic mutations in other families have been found and even more mutations are thought to exist. It is suspected that 25 percent of people with osteoarthritis have a specific gene mutation directly responsible for their osteoarthritis.
Is Arthritis All In The Family?
Researchers are not ready to declare genetics or familial factors as the sole cause of arthritis and related diseases. As more and more studies are done, the genetic factor seems more evident.
Why Me? Is It In The Genes?
Why does one person get arthritis as opposed to another person?
Why is a person afflicted with rheumatoid arthritis rather than osteoarthritis?
Why does one person have it much more severely than another person?
The endless "why" questions traverse the minds of people who suffer with arthritis, the people who care about them, the doctors who treat them, and the researchers who strive to find the answers.
Researchers and scientists are focused on what causes arthritis and what predisposes certain people to the disease. Many things have been considered as contributing to the cause of arthritis, including:
viruses
environmental factors
diet
other triggers
These and other factors have all been considered as relating to the cause.
zSB(3,3)
During the past few decades, genetics has become a more prominent area of arthritis research.
HLA-B27 Gene
A major genetic link was discovered back in the 1960's between the gene HLA-B27 and the spondyloarthropathies, a group of diseases affecting the spine and other joints.
90 percent of people with ankylosing spondylitis have the HLA-B27 gene.
7 percent of the general population have the HLA-B27 gene.
Scientists are working hard to find the other genes involved in the spondyloarthropathies and other rheumatic diseases.
During the decade following the discovery of HLA-B27, scientists found an association between carriers of the HLA-DR genes and increased risk of rheumatoid arthritis. It is believed that HLA-DR contributes to autoimmune disease, conditions caused by the immune system fighting the body it is supposed to protect. Other genes also are suspected of being major factors in the evolution of rheumatoid arthritis.
North American Rheumatoid Arthritis Consortium
The North American Rheumatoid Arthritis Consortium (which consists of 12 medical centers across the country, established and sponsored by the Arthritis Foundation, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Institute of Allergy and Infectious Diseases) has been analyzing clinical findings and genetic material from 1,000 pairs of siblings who both have rheumatoid arthritis. By testing 400 different genetic regions, researchers hope to identify specific genes associated with the rheumatoid arthritis.
HLA-DR4 Gene
The HLA-DR4 gene, which has been associated with rheumatoid arthritis, has also shown involvement inLyme disease. Lyme disease is caused by a microorganism which is transmitted to humans via deer ticks. Among the symptoms which can develop from Lyme disease are:
joint pain
inflammation
arthritis
People who have the disease more severely and do not respond well to the antibiotic treatment are more often found to have the HLA-DR4 gene. It has been theorized that once the microorganism moves to the joints, the immune response against it cross reacts with the person's own joint tissue in people who have the HLA-DR4 gene, leading to an autoimmune reaction.
Genetic Mutations
Other research has led to the discovery of a genetic mutation which causes osteoarthritis in some people. Several members of a family who had early onset osteoarthritis were found to have a genetic mutation in type II collagen. The mutation caused premature breakdown of joint cartilage in the affected family members. Since this discovery other genetic mutations in other families have been found and even more mutations are thought to exist. It is suspected that 25 percent of people with osteoarthritis have a specific gene mutation directly responsible for their osteoarthritis.
Is Arthritis All In The Family?
Researchers are not ready to declare genetics or familial factors as the sole cause of arthritis and related diseases. As more and more studies are done, the genetic factor seems more evident.
Genetics and hypertension
Genetic factors may contribute to an estimated thirty percent of cases of essential hypertension (high blood pressure of unknown cause). In the United States, high blood pressure occurs more frequently among African Americans than among white or Asian Americans. Adult African American men are most at risk for developing hypertension and cardiovascular diseases. The reason remains unknown, especially since non-American adult African men have very low occurrences of hypertension. A large percentage of people with essential hypertension have genetic abnormalities of their peripheral arteries (arterioles) — the small arteries that supply blood to the body's tissues. This genetic abnormality makes the walls of the arteries stiff so there is greater resistance to the blood flowing through them.
Genetics and kidney stones
A variety of factors can create the conditions that allow kidney stones to form. These factors include:
Some disturbances in the body's metabolism
Certain inherited defects
Abnormalities within the body
Sometimes, however, it's impossible to determine why a kidney stone has developed.
Each type of stone has its own requirements for formation.
Calcium stones form from the following:
Most calcium stones form for unknown reasons, although a genetic basis is suspected.
Certain foods can upset the balance of acid in the urine.
Cancer can cause the body to produce an abnormally large amount of parathyroid hormone, which regulates calcium levels in the body. High levels of this hormone can break down bone and releases too much calcium into the blood. As a result, calcium saturates the urine.
Uric acid stones form from the following:
Gout, the result of a genetic defect, is a disease that increases the body's production of uric acid. High levels of uric acid in the urine can cause stones to form.
An inherited tendency can lead to the development of this type of stone, although the specific reason is unknown.
Certain gastrointestinal conditions, such as ulcerative colitis, also can lead to the formation of uric acid stones.
Cystine stones can form from high amounts of cystine in the urine, the result of a rare in inherited abnormality.
Struvite stones may form in the kidney or bladder as a result of infection from certain bacteria.
Some disturbances in the body's metabolism
Certain inherited defects
Abnormalities within the body
Sometimes, however, it's impossible to determine why a kidney stone has developed.
Each type of stone has its own requirements for formation.
Calcium stones form from the following:
Most calcium stones form for unknown reasons, although a genetic basis is suspected.
Certain foods can upset the balance of acid in the urine.
Cancer can cause the body to produce an abnormally large amount of parathyroid hormone, which regulates calcium levels in the body. High levels of this hormone can break down bone and releases too much calcium into the blood. As a result, calcium saturates the urine.
Uric acid stones form from the following:
Gout, the result of a genetic defect, is a disease that increases the body's production of uric acid. High levels of uric acid in the urine can cause stones to form.
An inherited tendency can lead to the development of this type of stone, although the specific reason is unknown.
Certain gastrointestinal conditions, such as ulcerative colitis, also can lead to the formation of uric acid stones.
Cystine stones can form from high amounts of cystine in the urine, the result of a rare in inherited abnormality.
Struvite stones may form in the kidney or bladder as a result of infection from certain bacteria.
Genetics and diabetes
You've probably wondered how you got diabetes. You may worry that your children will get it too.
Unlike some traits, diabetes does not seem to be inherited in a simple pattern. Yet clearly, some people are born more likely to get diabetes than others.
What leads to diabetes?
Type 1 and type 2 diabetes have different causes. Yet two factors are important in both. First, you must inherit a predisposition to the disease. Second, something in your environment must trigger diabetes.
Genes alone are not enough. One proof of this is identical twins. Identical twins have identical genes. Yet when one twin has type 1 diabetes, the other gets the disease at most only half the time. When one twin has type 2 diabetes, the other's risk is at most 3 in 4.
Type 1 diabetes
In most cases of type 1 diabetes, people need to inherit risk factors from both parents. We think these factors must be more common in whites because whites have the highest rate of type 1 diabetes. Because most people who are at risk do not get diabetes, researchers want to find out what the environmental triggers are.
One trigger might be related to cold weather. Type 1 diabetes develops more often in winter than summer and is more common in places with cold climates. Another trigger might be viruses. Perhaps a virus that has only mild effects on most people triggers type 1 diabetes in others.
Early diet may also play a role. Type 1 diabetes is less common in people who were breastfed and in those who first ate solid foods at later ages.
In many people, the development of type 1 diabetes seems to take many years. In experiments that followed relatives of people with type 1 diabetes, researchers found that most of those who later got diabetes had certain autoantibodies in their blood for years before.
(Antibodies are proteins that destroy bacteria or viruses. Autoantibodies are antibodies 'gone bad,' which attack the body's own tissues.)
Type 2 diabetes
Type 2 diabetes has a stronger genetic basis than type 1, yet it also depends more on environmental factors. Sound confusing? What happens is that a family history of type 2 diabetes is one of the strongest risk factors for getting the disease but it only seems to matter in people living a Western lifestyle.
Americans and Europeans eat too much fat and too little carbohydrate and fiber, and they get too little exercise. Type 2 diabetes is common in people with these habits. The ethnic groups in the United States with the highest risk are African Americans, Mexican Americans, and Pima Indians.
In contrast, people who live in areas that have not become Westernized tend not to get type 2 diabetes, no matter how high their genetic risk.
Obesity is a strong risk factor for type 2 diabetes. Obesity is most risky for young people and for people who have been obese for a long time.
Gestational diabetes is more of a puzzle. Women who get diabetes while they are pregnant are more likely to have a family history of diabetes, especially on their mothers' side. But as in other forms of diabetes, nongenetic factors play a role. Older mothers and overweight women are more likely to get gestational diabetes.
Unlike some traits, diabetes does not seem to be inherited in a simple pattern. Yet clearly, some people are born more likely to get diabetes than others.
What leads to diabetes?
Type 1 and type 2 diabetes have different causes. Yet two factors are important in both. First, you must inherit a predisposition to the disease. Second, something in your environment must trigger diabetes.
Genes alone are not enough. One proof of this is identical twins. Identical twins have identical genes. Yet when one twin has type 1 diabetes, the other gets the disease at most only half the time. When one twin has type 2 diabetes, the other's risk is at most 3 in 4.
Type 1 diabetes
In most cases of type 1 diabetes, people need to inherit risk factors from both parents. We think these factors must be more common in whites because whites have the highest rate of type 1 diabetes. Because most people who are at risk do not get diabetes, researchers want to find out what the environmental triggers are.
One trigger might be related to cold weather. Type 1 diabetes develops more often in winter than summer and is more common in places with cold climates. Another trigger might be viruses. Perhaps a virus that has only mild effects on most people triggers type 1 diabetes in others.
Early diet may also play a role. Type 1 diabetes is less common in people who were breastfed and in those who first ate solid foods at later ages.
In many people, the development of type 1 diabetes seems to take many years. In experiments that followed relatives of people with type 1 diabetes, researchers found that most of those who later got diabetes had certain autoantibodies in their blood for years before.
(Antibodies are proteins that destroy bacteria or viruses. Autoantibodies are antibodies 'gone bad,' which attack the body's own tissues.)
Type 2 diabetes
Type 2 diabetes has a stronger genetic basis than type 1, yet it also depends more on environmental factors. Sound confusing? What happens is that a family history of type 2 diabetes is one of the strongest risk factors for getting the disease but it only seems to matter in people living a Western lifestyle.
Americans and Europeans eat too much fat and too little carbohydrate and fiber, and they get too little exercise. Type 2 diabetes is common in people with these habits. The ethnic groups in the United States with the highest risk are African Americans, Mexican Americans, and Pima Indians.
In contrast, people who live in areas that have not become Westernized tend not to get type 2 diabetes, no matter how high their genetic risk.
Obesity is a strong risk factor for type 2 diabetes. Obesity is most risky for young people and for people who have been obese for a long time.
Gestational diabetes is more of a puzzle. Women who get diabetes while they are pregnant are more likely to have a family history of diabetes, especially on their mothers' side. But as in other forms of diabetes, nongenetic factors play a role. Older mothers and overweight women are more likely to get gestational diabetes.
Genetics and asthma
While the inheritable nature of allergic conditions has been known for more than a century, it is clearly a hot topic with a rapidly proliferating literature.
Allergic conditions each involve complex cellular and molecular interactions that converge to produce common mechanisms of disease. As a result, new studies in a range of allergic conditions continue to highlight novel genes and pathways that may be critical in particular clinical situations.
Of the various allergic conditions, asthma has attracted most attention in terms of defining its precise genetic basis and serves as a model for considering progress. Asthma is a common heterogeneous respiratory condition characterised by several pathological features, including chronic inflammation of the airways, reversible bronchorestriction, excessive mucous production and airway hyperreactivity.
Affected individuals generally present with chest tightness, wheezing and breathlessness. Asthma is a complex disease and poses a number of research challenges in terms of identifying susceptibility determinants, including the changing nature of the disease throughout life.
The long-held clinical tenet in asthma is that the disease arises in a subset of individuals with genetic susceptibility, after exposure to an environmental trigger. This is based on early epidemiological and twin studies which confirmed a role for both genetic and environmental influences.
Early findings included a four- to five-fold increase in asthma prevalence in first-order relatives of affected individuals and greater concordance of the disease in monozygotic twins relative to dizygotic twins.
As is the case with many other complex diseases, this clear evidence of genetic influence coupled with new technological advances raised the hope of relatively rapid advances in gene-based diagnostic and therapeutic approaches for asthma. For example, gene arrays predicting asthma risk with reasonable precision early in life seemed realistic within a limited timeframe. The reality is, however, a little more measured.
Given the high prevalence of the condition, interest in identifying genes involved in asthma susceptibility has been necessarily intense, with many candidate genes identified from both guided association studies and exploratory whole genome linkage analyses. Genes so far identified encode a diverse array of molecules including ion channels, cytokines, receptors, enzymes and transporters.
Despite the high level of genetic scrutiny in asthma and plethora of candidate genes, evidence relating to the majority of genes remains patchy and in many cases, controversial. Many factors have been identified as contributing to this lack of clarity including a range of methodological issues and the heterogeneous nature of the disease itself.
Two major issues loom in most studies of asthma genetics. Firstly, many genetic variants associated with asthma appear to be only important in a relatively small number of individuals. Secondly, considerable difficulty has been experienced in replicating studies across ethnically or geographically diverse populations. As a result, there is no genetic test available that would reliably predict asthma risk in a clinically meaningful way. There is a major upside, however, to all of this genetic trawling, with exciting new insights into the biological pathways of asthma that are likely to lead to novel therapeutic approaches.
Allergic conditions each involve complex cellular and molecular interactions that converge to produce common mechanisms of disease. As a result, new studies in a range of allergic conditions continue to highlight novel genes and pathways that may be critical in particular clinical situations.
Of the various allergic conditions, asthma has attracted most attention in terms of defining its precise genetic basis and serves as a model for considering progress. Asthma is a common heterogeneous respiratory condition characterised by several pathological features, including chronic inflammation of the airways, reversible bronchorestriction, excessive mucous production and airway hyperreactivity.
Affected individuals generally present with chest tightness, wheezing and breathlessness. Asthma is a complex disease and poses a number of research challenges in terms of identifying susceptibility determinants, including the changing nature of the disease throughout life.
The long-held clinical tenet in asthma is that the disease arises in a subset of individuals with genetic susceptibility, after exposure to an environmental trigger. This is based on early epidemiological and twin studies which confirmed a role for both genetic and environmental influences.
Early findings included a four- to five-fold increase in asthma prevalence in first-order relatives of affected individuals and greater concordance of the disease in monozygotic twins relative to dizygotic twins.
As is the case with many other complex diseases, this clear evidence of genetic influence coupled with new technological advances raised the hope of relatively rapid advances in gene-based diagnostic and therapeutic approaches for asthma. For example, gene arrays predicting asthma risk with reasonable precision early in life seemed realistic within a limited timeframe. The reality is, however, a little more measured.
Given the high prevalence of the condition, interest in identifying genes involved in asthma susceptibility has been necessarily intense, with many candidate genes identified from both guided association studies and exploratory whole genome linkage analyses. Genes so far identified encode a diverse array of molecules including ion channels, cytokines, receptors, enzymes and transporters.
Despite the high level of genetic scrutiny in asthma and plethora of candidate genes, evidence relating to the majority of genes remains patchy and in many cases, controversial. Many factors have been identified as contributing to this lack of clarity including a range of methodological issues and the heterogeneous nature of the disease itself.
Two major issues loom in most studies of asthma genetics. Firstly, many genetic variants associated with asthma appear to be only important in a relatively small number of individuals. Secondly, considerable difficulty has been experienced in replicating studies across ethnically or geographically diverse populations. As a result, there is no genetic test available that would reliably predict asthma risk in a clinically meaningful way. There is a major upside, however, to all of this genetic trawling, with exciting new insights into the biological pathways of asthma that are likely to lead to novel therapeutic approaches.
Genetic and heart disease
A tendency toward heart disease or fatty buildups in arteries seems to be hereditary. That means children of parents with heart and blood vessel diseases may be more likely to develop them. Race is also a factor. African Americans have higher risk of developing high blood pressure. This makes their risk of heart disease and stroke greater.
A family history of diabetes (di"ah-BE'teez or di"ah-BE'tis), gout, high blood pressure or high blood cholesterol also increases the risk of heart disease.
A number of genes have been reported to be associated with heart disease, stroke and high blood pressure in large population-based studies. However, the impact of each individual gene on an individual person is not fully understood.
A person with a congenital heart defect is slightly more likely than the general public to have a baby with a congenital heart defect. Researchers are now identifying genes responsible for causing some of these defects.
Even though you can't change your genetic makeup, you can reduce your risk by adopting a healthier lifestyle that includes physical activity, a healthy diet, and avoiding tobacco.
You can learn more about your family history by asking questions, talking at family gatherings, and looking at family medical records, if possible. Try to learn about the medical history of your grandparents, parents, aunts and uncles, nieces and nephews, siblings, and children. You should try to find out the following:
Major medical conditions and causes of death.
Age of disease onset and age at death, and
Ethnic background.
Please share your family history information with your doctor. Your doctor will:
Assess your disease risk.
Recommend lifestyle changes to help prevent disease, and
Prescribe laboratory or clinical tests to detect disease early.
A family history of diabetes (di"ah-BE'teez or di"ah-BE'tis), gout, high blood pressure or high blood cholesterol also increases the risk of heart disease.
A number of genes have been reported to be associated with heart disease, stroke and high blood pressure in large population-based studies. However, the impact of each individual gene on an individual person is not fully understood.
A person with a congenital heart defect is slightly more likely than the general public to have a baby with a congenital heart defect. Researchers are now identifying genes responsible for causing some of these defects.
Even though you can't change your genetic makeup, you can reduce your risk by adopting a healthier lifestyle that includes physical activity, a healthy diet, and avoiding tobacco.
You can learn more about your family history by asking questions, talking at family gatherings, and looking at family medical records, if possible. Try to learn about the medical history of your grandparents, parents, aunts and uncles, nieces and nephews, siblings, and children. You should try to find out the following:
Major medical conditions and causes of death.
Age of disease onset and age at death, and
Ethnic background.
Please share your family history information with your doctor. Your doctor will:
Assess your disease risk.
Recommend lifestyle changes to help prevent disease, and
Prescribe laboratory or clinical tests to detect disease early.
Genetics and stroke
Having a family history of stroke increases the risk of stroke, but only by a small amount.
If many of your blood relatives have had stroke or heart attacks then it would be sensible to get your doctor to give you a check-up. In particular it would be important to check your blood pressure and cholesterol level. Certain racial groups are at greater risk of stroke than others; in particular people from West Africa and the Caribbean have twice the rate of stroke than Caucasians. Part of this difference may represent genetic factors.
If many of your blood relatives have had stroke or heart attacks then it would be sensible to get your doctor to give you a check-up. In particular it would be important to check your blood pressure and cholesterol level. Certain racial groups are at greater risk of stroke than others; in particular people from West Africa and the Caribbean have twice the rate of stroke than Caucasians. Part of this difference may represent genetic factors.
Tuesday, 20 November 2007
My project (13)
A critical part of the MNK and WND physiological response are their ability to change intracellular localisation in response to copper levels, relocating to sites where copper transport is required.
Under basal physiological copper levels, MNK and WND concentrate within the trans Golgi network (TGN) region, where they are postulated to pump copper into the TGN lumen for incorporatio into proteins on the secretory pathway.
When copper levels are raised, MNK has been reported to traffic to the plasma membrane and to the basolateral membrane in some polarized cell.
In response to elevated copper levels WND traffics to sub apical vesicles in some polarized cell lines, and has also been deonstrated partially at the apical membrane.
When intracellular copper levels are reduced, both transporters return via an endocytic route to the TGN.
Under basal physiological copper levels, MNK and WND concentrate within the trans Golgi network (TGN) region, where they are postulated to pump copper into the TGN lumen for incorporatio into proteins on the secretory pathway.
When copper levels are raised, MNK has been reported to traffic to the plasma membrane and to the basolateral membrane in some polarized cell.
In response to elevated copper levels WND traffics to sub apical vesicles in some polarized cell lines, and has also been deonstrated partially at the apical membrane.
When intracellular copper levels are reduced, both transporters return via an endocytic route to the TGN.
My project (12)
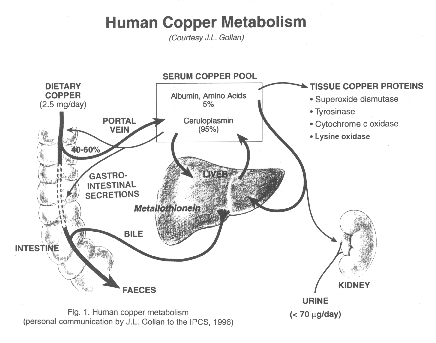
Human copper metabolism
Ingested copper is absorp from the gut 40-50%.
Copper bind to protein (mostly ceruloplasmin 95%).
Copper is transported to liver. Copper is secreted to bile and excreted to the gut again in feces
Subscribe to:
Comments (Atom)